Cytokine Signaling in Multiple Sclerosis and Its Therapeutic Applications


Abstract
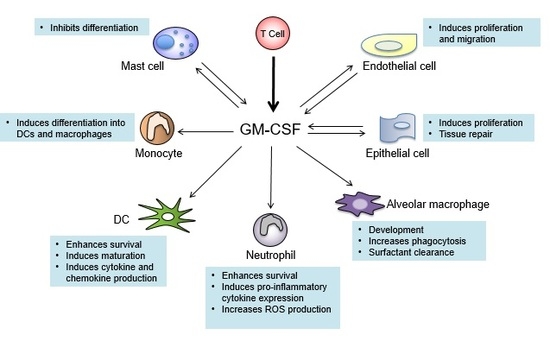
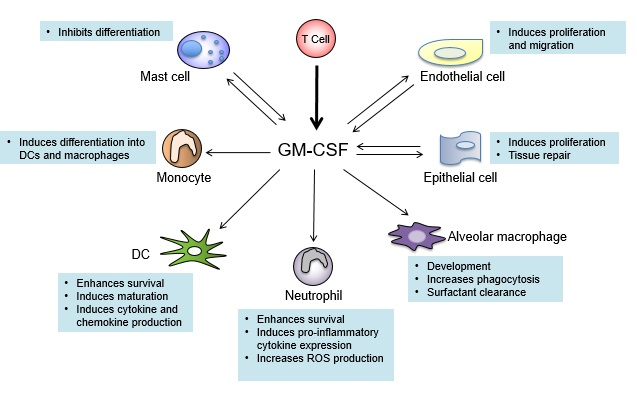
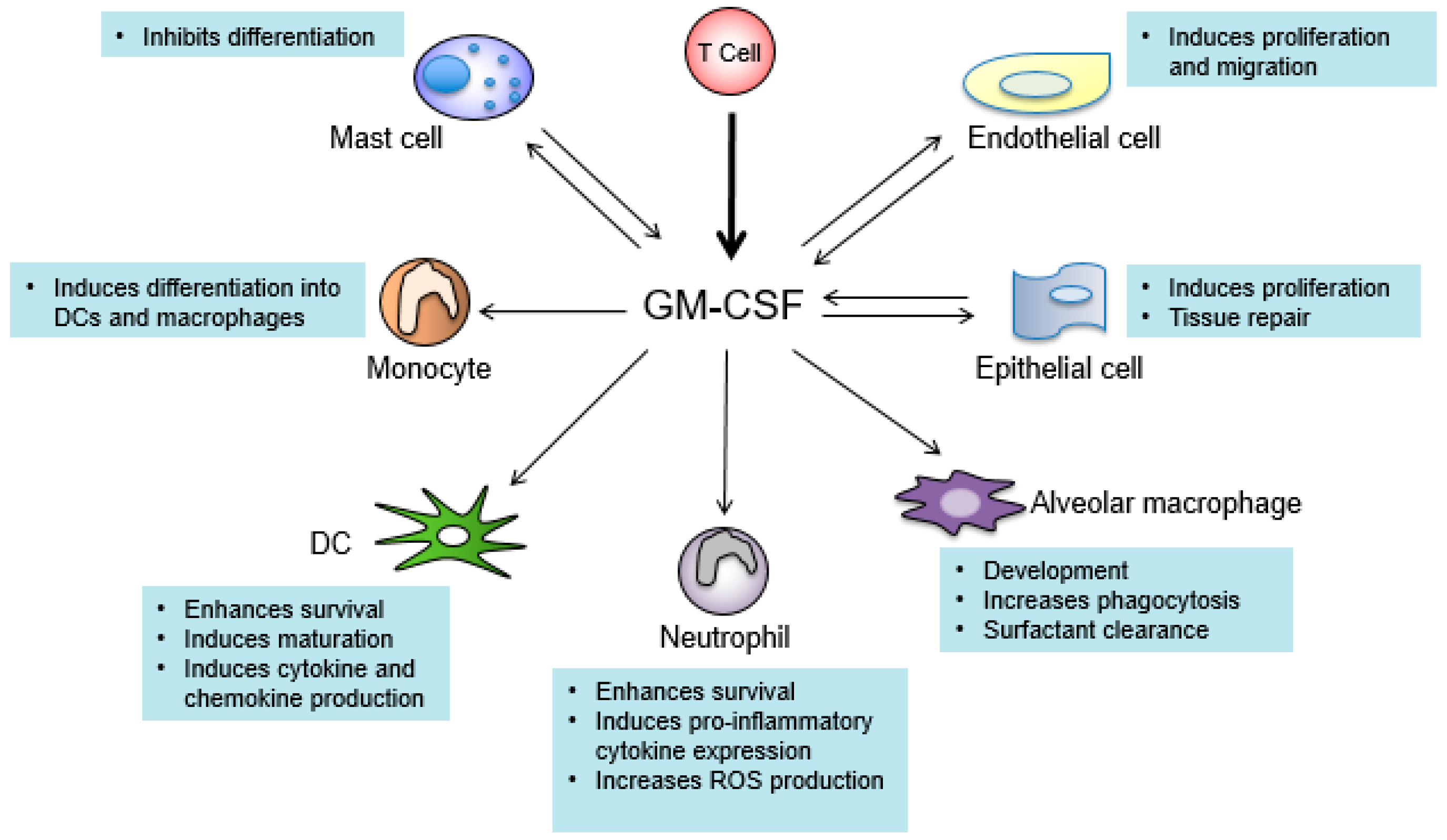
:
1. Introduction
2. Diagnosis and Clinical Courses of Multiple Sclerosis
3. Multiple Sclerosis as an Immune-Mediated Disease
4. Experimental Autoimmune Encephalomyelitis Is a Valuable Model for Multiple Sclerosis Research
5. Cytokines Involved in the Pathogenesis of Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis
6. The IL-23-IL17 Axis in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis
7. GM-CSF Connects the Priming Autoreactive CD4+ T Cells to the Effector Myeloid Cells
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Goldenberg, M.M. Multiple sclerosis review. Pharm. Ther. 2012, 37, 175–184. [Google Scholar]
- Bove, R.; Chitnis, T. Sexual disparities in the incidence and course of MS. Clin. Immunol. 2013, 149, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Castrop, F.; Haslinger, B.; Hemmer, B.; Buck, D. Review of the pharmacoeconomics of early treatment of multiple sclerosis using interferon beta. Neuropsychiatr. Dis. Treat. 2013, 9, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Tullman, M.J. Overview of the epidemiology, diagnosis, and disease progression associated with multiple sclerosis. Am. J. Manag. Care 2013, 19, S15–S20. [Google Scholar] [PubMed]
- Naci, H.; Fleurence, R.; Birt, J.; Duhig, A. Economic burden of multiple sclerosis: A systematic review of the literature. Pharmacoeconomics 2010, 28, 363–379. [Google Scholar] [CrossRef] [PubMed]
- Loma, I.; Heyman, R. Multiple sclerosis: Pathogenesis and treatment. Curr. Neuropharmacol. 2011, 9, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Kister, I.; Bacon, T.E.; Chamot, E.; Salter, A.R.; Cutter, G.R.; Kalina, J.T.; Herbert, J. Natural history of multiple sclerosis symptoms. Int. J. MS Care 2013, 15, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Filippi, M.; Rocca, M.A.; Ciccarelli, O.; De Stefano, N.; Evangelou, N.; Kappos, L.; Rovira, A.; Sastre-Garriga, J.; Tintore, M.; Frederiksen, J.L.; et al. MRI criteria for the diagnosis of multiple sclerosis: Magnims consensus guidelines. Lancet Neurol. 2016, 15, 292–303. [Google Scholar] [CrossRef]
- Wattjes, M.P.; Steenwijk, M.D.; Stangel, M. MRI in the diagnosis and monitoring of multiple sclerosis: An update. Clin. Neuroradiol. 2015, 25, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Polman, C.H.; Reingold, S.C.; Banwell, B.; Clanet, M.; Cohen, J.A.; Filippi, M.; Fujihara, K.; Havrdova, E.; Hutchinson, M.; Kappos, L.; et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the mcdonald criteria. Ann. Neurol. 2011, 69, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sorensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Fitzner, D.; Simons, M. Chronic progressive multiple sclerosis—Pathogenesis of neurodegeneration and therapeutic strategies. Curr. Neuropharmacol. 2010, 8, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Applebee, A.; Panitch, H. Early stage and long term treatment of multiple sclerosis with interferon-beta. Biologics 2009, 3, 257–271. [Google Scholar] [PubMed]
- Andersson, P.B.; Waubant, E.; Gee, L.; Goodkin, D.E. Multiple sclerosis that is progressive from the time of onset: Clinical characteristics and progression of disability. Arch. Neurol. 1999, 56, 1138–1142. [Google Scholar] [CrossRef] [PubMed]
- Gold, R.; Wolinsky, J.S.; Amato, M.P.; Comi, G. Evolving expectations around early management of multiple sclerosis. Ther. Adv. Neurol. Disord. 2010, 3, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Wolinsky, J.S.; Group, P.R.S. The diagnosis of primary progressive multiple sclerosis. J. Neurol. Sci. 2003, 206, 145–152. [Google Scholar] [CrossRef]
- Cottrell, D.A.; Kremenchutzky, M.; Rice, G.P.; Hader, W.; Baskerville, J.; Ebers, G.C. The natural history of multiple sclerosis: A geographically based study. 6. Applications to planning and interpretation of clinical therapeutic trials in primary progressive multiple sclerosis. Brain 1999, 122, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Richard-Miceli, C.; Criswell, L.A. Emerging patterns of genetic overlap across autoimmune disorders. Genome Med. 2012, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Bashinskaya, V.V.; Kulakova, O.G.; Boyko, A.N.; Favorov, A.V.; Favorova, O.O. A review of genome-wide association studies for multiple sclerosis: Classical and hypothesis-driven approaches. Hum. Genet. 2015, 134, 1143–1162. [Google Scholar] [CrossRef] [PubMed]
- Didonna, A.; Oksenberg, J.R. Genetic determinants of risk and progression in multiple sclerosis. Clin. Chim. Acta 2015, 449, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.F.; Alvarez, E. The immunopathophysiology of multiple sclerosis. Neurol. Clin. 2011, 29, 257–278. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.K.; Yong, V.W. Myeloid cells—Targets of medication in multiple sclerosis. Nat. Rev. Neurol. 2016, 12, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Goods, B.A.; Raddassi, K.; Nepom, G.T.; Kwok, W.W.; Love, J.C.; Hafler, D.A. Functional inflammatory profiles distinguish myelin-reactive T cells from patients with multiple sclerosis. Sci. Transl. Med. 2015, 7, 287ra274. [Google Scholar] [CrossRef] [PubMed]
- Adorini, L. Tolerogenic dendritic cells induced by vitamin d receptor ligands enhance regulatory T cells inhibiting autoimmune diabetes. Ann. N. Y. Acad. Sci. 2003, 987, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Chen, J.; Zheng, C.; Wu, J.; Cheng, Y.; Zhu, S.; Lin, C.; Cao, Q.; Zhu, J.; Jin, T. 1,25-dihydroxyvitamin d3-induced dendritic cells suppress experimental autoimmune encephalomyelitis by increasing proportions of the regulatory lymphocytes and reducing T helper type 1 and type 17 cells. Immunology 2017, 152, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, D.S.; Hygino, J.; Ferreira, T.B.; Kasahara, T.M.; Barros, P.O.; Monteiro, C.; Oliveira, A.; Tavares, F.; Vasconcelos, C.C.; Alvarenga, R.; et al. Vitamin d modulates different IL-17-secreting T cell subsets in multiple sclerosis patients. J. Neuroimmunol. 2016, 299, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Bermudez-Morales, V.H.; Fierros, G.; Lopez, R.L.; Martinez-Nava, G.; Flores-Aldana, M.; Flores-Rivera, J.; Hernandez-Giron, C. Vitamin d receptor gene polymorphisms are associated with multiple sclerosis in mexican adults. J. Neuroimmunol. 2017, 306, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Cierny, D.; Michalik, J.; Skerenova, M.; Kantorova, E.; Sivak, S.; Javor, J.; Kurca, E.; Dobrota, D.; Lehotsky, J. Apai, Bsmi and Taqi VDR gene polymorphisms in association with multiple sclerosis in Slovaks. Neurol. Res. 2016, 38, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.M.; Hughes, A.M.; Lay, M.L.; Ponsonby, A.L.; Dwyer, D.E.; Taylor, B.V.; Pender, M.P. Epstein-barr virus and multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 1142–1148. [Google Scholar] [CrossRef] [PubMed]
- Pender, M.P. The essential role of Epstein-Barr virus in the pathogenesis of multiple sclerosis. Neuroscientist 2011, 17, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Odumade, O.A.; Hogquist, K.A.; Balfour, H.H., Jr. Progress and problems in understanding and managing primary Epstein-Barr virus infections. Clin. Microbiol. Rev. 2011, 24, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Lunemann, J.D.; Kamradt, T.; Martin, R.; Munz, C. Epstein-barr virus: Environmental trigger of multiple sclerosis? J. Virol. 2007, 81, 6777–6784. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Hafler, D.A.; Lucchinetti, C.F. Multiple sclerosis-a quiet revolution. Nat. Rev. Neurol. 2015, 11, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.P.; Hartung, H.P.; Calabresi, P.A. Anti-alpha4 integrin therapy for multiple sclerosis: Mechanisms and rationale. Neurology 2005, 64, 1336–1342. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Billich, A.; Baumruker, T.; Heining, P.; Schmouder, R.; Francis, G.; Aradhye, S.; Burtin, P. Fingolimod (fty720): Discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Discov. 2010, 9, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.A.; Martin, R.; Jacobson, S. Chemokines in chronic progressive neurological diseases: Htlv-1 associated myelopathy and multiple sclerosis. J. Neurovirol. 1999, 5, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Brandstadter, R.; Katz Sand, I. The use of natalizumab for multiple sclerosis. Neuropsychiatr. Dis. Treat. 2017, 13, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef] [PubMed]
- Rangachari, M.; Kuchroo, V.K. Using eae to better understand principles of immune function and autoimmune pathology. J. Autoimmun. 2013, 45, 31–39. [Google Scholar] [CrossRef] [PubMed]
- McRae, B.L.; Vanderlugt, C.L.; Dal Canto, M.C.; Miller, S.D. Functional evidence for epitope spreading in the relapsing pathology of experimental autoimmune encephalomyelitis. J. Exp. Med. 1995, 182, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Whitham, R.H.; Bourdette, D.N.; Hashim, G.A.; Herndon, R.M.; Ilg, R.C.; Vandenbark, A.A.; Offner, H. Lymphocytes from SJL/J mice immunized with spinal cord respond selectively to a peptide of proteolipid protein and transfer relapsing demyelinating experimental autoimmune encephalomyelitis. J. Immunol. 1991, 146, 101–107. [Google Scholar] [PubMed]
- Mendel, I.; Kerlero de Rosbo, N.; Ben-Nun, A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2B mice: Fine specificity and T cell receptor V beta expression of encephalitogenic t cells. Eur. J. Immunol. 1995, 25, 1951–1959. [Google Scholar] [CrossRef] [PubMed]
- Kerfoot, S.M.; Long, E.M.; Hickey, M.J.; Andonegui, G.; Lapointe, B.M.; Zanardo, R.C.; Bonder, C.; James, W.G.; Robbins, S.M.; Kubes, P. Tlr4 contributes to disease-inducing mechanisms resulting in central nervous system autoimmune disease. J. Immunol. 2004, 173, 7070–7077. [Google Scholar] [CrossRef] [PubMed]
- Ronchi, F.; Basso, C.; Preite, S.; Reboldi, A.; Baumjohann, D.; Perlini, L.; Lanzavecchia, A.; Sallusto, F. Experimental priming of encephalitogenic Th1/Th17 cells requires pertussis toxin-driven IL-1beta production by myeloid cells. Nat. Commun. 2016, 7, 11541. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Pagany, M.; Weiner, H.L.; Linington, C.; Sobel, R.A.; Kuchroo, V.K. Myelin oligodendrocyte glycoprotein-specific t cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J. Exp. Med. 2003, 197, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Stromnes, I.M.; Goverman, J.M. Passive induction of experimental allergic encephalomyelitis. Nat. Protoc. 2006, 1, 1952–1960. [Google Scholar] [CrossRef] [PubMed]
- Cua, D.J.; Sherlock, J.; Chen, Y.; Murphy, C.A.; Joyce, B.; Seymour, B.; Lucian, L.; To, W.; Kwan, S.; Churakova, T.; et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 2003, 421, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of cd4 t cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Teitelbaum, D.; Meshorer, A.; Hirshfeld, T.; Arnon, R.; Sela, M. Suppression of experimental allergic encephalomyelitis by a synthetic polypeptide. Eur. J. Immunol. 1971, 1, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Teitelbaum, D.; Webb, C.; Meshorer, A.; Arnon, R.; Sela, M. Protection against experimental allergic encephalomyelitis. Nature 1972, 240, 564–566. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, M.; Avolio, C.; Livrea, P.; Trojano, M. Glatiramer acetate in multiple sclerosis: A review. CNS Drug Rev. 2007, 13, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.P.; Brooks, B.R.; Cohen, J.A.; Ford, C.C.; Goldstein, J.; Lisak, R.P.; Myers, L.W.; Panitch, H.S.; Rose, J.W.; Schiffer, R.B. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: Results of a phase iii multicenter, double-blind placebo-controlled trial. The copolymer 1 multiple sclerosis study group. Neurology 1995, 45, 1268–1276. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.P.; Brooks, B.R.; Cohen, J.A.; Ford, C.C.; Goldstein, J.; Lisak, R.P.; Myers, L.W.; Panitch, H.S.; Rose, J.W.; Schiffer, R.B.; et al. Extended use of glatiramer acetate (copaxone) is well tolerated and maintains its clinical effect on multiple sclerosis relapse rate and degree of disability. Copolymer 1 multiple sclerosis study group. Neurology 1998, 50, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Jee, Y.; Piao, W.H.; Liu, R.; Bai, X.F.; Rhodes, S.; Rodebaugh, R.; Campagnolo, D.I.; Shi, F.D.; Vollmer, T.L. Cd4(+)cd25(+) regulatory t cells contribute to the therapeutic effects of glatiramer acetate in experimental autoimmune encephalomyelitis. Clin. Immunol. 2007, 125, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, R.; Eilam, R.; Stock, A.; Vainshtein, A.; Shezen, E.; Gal, H.; Friedman, N.; Arnon, R. Glatiramer acetate reduces th-17 inflammation and induces regulatory T-cells in the cns of mice with relapsing-remitting or chronic eae. J. Neuroimmunol. 2010, 225, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Begum-Haque, S.; Sharma, A.; Kasper, I.R.; Foureau, D.M.; Mielcarz, D.W.; Haque, A.; Kasper, L.H. Downregulation of IL-17 and IL-6 in the central nervous system by glatiramer acetate in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2008, 204, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, R.; Meshorer, A.; Sela, M.; Arnon, R. Oral treatment of mice with copolymer 1 (glatiramer acetate) results in the accumulation of specific th2 cells in the central nervous system. J. Neuroimmunol. 2002, 126, 58–68. [Google Scholar] [CrossRef]
- Toker, A.; Slaney, C.Y.; Backstrom, B.T.; Harper, J.L. Glatiramer acetate treatment directly targets cd11b(+)ly6g(−) monocytes and enhances the suppression of autoreactive t cells in experimental autoimmune encephalomyelitis. Scand. J. Immunol. 2011, 74, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, R.; Herschkovitz, A.; Eilam, R.; Blumberg-Hazan, M.; Sela, M.; Bruck, W.; Arnon, R. Demyelination arrest and remyelination induced by glatiramer acetate treatment of experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2008, 105, 11358–11363. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, R.; Eilam, R.; Domev, H.; Labunskay, G.; Sela, M.; Arnon, R. The immunomodulator glatiramer acetate augments the expression of neurotrophic factors in brains of experimental autoimmune encephalomyelitis mice. Proc. Natl. Acad. Sci. USA 2005, 102, 19045–19050. [Google Scholar] [CrossRef] [PubMed]
- Yednock, T.A.; Cannon, C.; Fritz, L.C.; Sanchez-Madrid, F.; Steinman, L.; Karin, N. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature 1992, 356, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Baron, J.L.; Madri, J.A.; Ruddle, N.H.; Hashim, G.; Janeway, C.A., Jr. Surface expression of alpha 4 integrin by cd4 t cells is required for their entry into brain parenchyma. J. Exp. Med. 1993, 177, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.H.; Khan, O.A.; Sheremata, W.A.; Blumhardt, L.D.; Rice, G.P.; Libonati, M.A.; Willmer-Hulme, A.J.; Dalton, C.M.; Miszkiel, K.A.; O’Connor, P.W.; et al. A controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 2003, 348, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Polman, C.H.; O’Connor, P.W.; Havrdova, E.; Hutchinson, M.; Kappos, L.; Miller, D.H.; Phillips, J.T.; Lublin, F.D.; Giovannoni, G.; Wajgt, A.; et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 2006, 354, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Bittner, S.; Afzali, A.M.; Wiendl, H.; Meuth, S.G. Myelin oligodendrocyte glycoprotein (mog35–55) induced experimental autoimmune encephalomyelitis (EAE) in c57bl/6 mice. J. Vis. Exp. 2014. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.M.; Lalor, S.J.; Sweeney, C.M.; Tubridy, N.; Mills, K.H. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin. Exp. Immunol. 2010, 162, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hohlfeld, R.; Dornmair, K.; Meinl, E.; Wekerle, H. The search for the target antigens of multiple sclerosis, part 2: Cd8+ T cells, b cells, and antibodies in the focus of reverse-translational research. Lancet Neurol. 2016, 15, 317–331. [Google Scholar] [CrossRef]
- Goverman, J. Autoimmune T cell responses in the central nervous system. Nat. Rev. Immunol. 2009, 9, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Langer-Gould, A.; Atlas, S.W.; Green, A.J.; Bollen, A.W.; Pelletier, D. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N. Engl. J. Med. 2005, 353, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.R. Natalizumab and progressive multifocal leucoencephalopathy. Ann. Rheum. Dis. 2006, 65, iii48–iii53. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, F.; Kajioka, J.; Miyamura, T. Prevalence rate and age of acquisition of antibodies against JC virus and BK virus in human sera. Microbiol. Immunol. 1982, 26, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Planas, R.; Martin, R.; Sospedra, M. Long-term safety and efficacy of natalizumab in relapsing-remitting multiple sclerosis: Impact on quality of life. Patient Relat. Outcome Meas. 2014, 5, 25–33. [Google Scholar] [PubMed]
- Owens, G.P.; Gilden, D.; Burgoon, M.P.; Yu, X.; Bennett, J.L. Viruses and multiple sclerosis. Neuroscientist 2011, 17, 659–676. [Google Scholar] [CrossRef] [PubMed]
- Pevear, D.C.; Calenoff, M.; Rozhon, E.; Lipton, H.L. Analysis of the complete nucleotide sequence of the picornavirus theiler’s murine encephalomyelitis virus indicates that it is closely related to cardioviruses. J. Virol. 1987, 61, 1507–1516. [Google Scholar] [PubMed]
- Oleszak, E.L.; Chang, J.R.; Friedman, H.; Katsetos, C.D.; Platsoucas, C.D. Theiler’s virus infection: A model for multiple sclerosis. Clin. Microbiol. Rev. 2004, 17, 174–207. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, I.; Fujinami, R.S. Neuropathogenesis of theiler’s murine encephalomyelitis virus infection, an animal model for multiple sclerosis. J. Neuroimmune Pharmacol. 2010, 5, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, I.; Iwasaki, Y.; Terunuma, H.; Sako, K.; Ohara, Y. A comparative study of acute and chronic diseases induced by two subgroups of theiler’s murine encephalomyelitis virus. Acta Neuropathol. 1996, 91, 595–602. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.P.; Richards, M.H.; Miller, S.D. Mouse models of multiple sclerosis: Experimental autoimmune encephalomyelitis and theiler’s virus-induced demyelinating disease. Methods Mol. Biol. 2012, 900, 381–401. [Google Scholar] [PubMed]
- Denic, A.; Johnson, A.J.; Bieber, A.J.; Warrington, A.E.; Rodriguez, M.; Pirko, I. The relevance of animal models in multiple sclerosis research. Pathophysiology 2011, 18, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Praet, J.; Guglielmetti, C.; Berneman, Z.; Van der Linden, A.; Ponsaerts, P. Cellular and molecular neuropathology of the cuprizone mouse model: Clinical relevance for multiple sclerosis. Neurosci. Biobehav. Rev. 2014, 47, 485–505. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; Bradl, M. Multiple sclerosis: Experimental models and reality. Acta Neuropathol. 2017, 133, 223–244. [Google Scholar] [CrossRef] [PubMed]
- Blakemore, W.F.; Franklin, R.J. Remyelination in experimental models of toxin-induced demyelination. Adv. Mult. Scler. Expe. Demyelinating Dis. 2008, 318, 193–212. [Google Scholar]
- Torkildsen, O.; Brunborg, L.A.; Myhr, K.M.; Bo, L. The cuprizone model for demyelination. Acta Neurol. Scand. 2008, 188, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Gudi, V.; Gingele, S.; Skripuletz, T.; Stangel, M. Glial response during cuprizone-induced DE- and remyelination in the CNS: Lessons learned. Front. Cell. Neurosci. 2014, 8, 73. [Google Scholar] [CrossRef] [PubMed]
- Van Engelen, B.G.; Pavelko, K.D.; Rodriguez, M. Enhancement of central nervous system remyelination in immune and non-immune experimental models of demyelination. Mult. Scler. J. 1997, 3, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Engelhardt, B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 2012, 12, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Sharief, M.K.; Hentges, R. Association between tumor necrosis factor-alpha and disease progression in patients with multiple sclerosis. N. Engl. J. Med. 1991, 325, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Maimone, D.; Gregory, S.; Arnason, B.G.; Reder, A.T. Cytokine levels in the cerebrospinal fluid and serum of patients with multiple sclerosis. J. Neuroimmunol. 1991, 32, 67–74. [Google Scholar] [CrossRef]
- Ruddle, N.H.; Bergman, C.M.; McGrath, K.M.; Lingenheld, E.G.; Grunnet, M.L.; Padula, S.J.; Clark, R.B. An antibody to lymphotoxin and tumor necrosis factor prevents transfer of experimental allergic encephalomyelitis. J. Exp. Med. 1990, 172, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.R.; Zaczynska, E.; Katsetos, C.D.; Platsoucas, C.D.; Oleszak, E.L. Differential expression of TGF-beta, IL-2, and other cytokines in the cns of theiler’s murine encephalomyelitis virus-infected susceptible and resistant strains of mice. Virology 2000, 278, 346–360. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Koh, C.S.; Yahikozawa, H.; Yanagisawa, N.; Yagita, H.; Ishihara, Y.; Kim, B.S. The level of tumor necrosis factor-alpha producing cells in the spinal cord correlates with the degree of theiler’s murine encephalomyelitis virus-induced demyelinating disease. Int. Immunol. 1996, 8, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- TNF neutralization in MS: Results of a randomized, placebo-controlled multicenter study. The lenercept multiple sclerosis study group and the university of british columbia MS/MRI analysis group. Neurology 1999, 53, 457–465.
- Figiel, I. Pro-inflammatory cytokine TNF-alpha as a neuroprotective agent in the brain. Acta Neurobiol. Exp. (Wars) 2008, 68, 526–534. [Google Scholar] [PubMed]
- Jacobs, L.; O’Malley, J.; Freeman, A.; Ekes, R. Intrathecal interferon reduces exacerbations of multiple sclerosis. Science 1981, 214, 1026–1028. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, L.; Salazar, A.M.; Herndon, R.; Reese, P.A.; Freeman, A.; Josefowicz, R.; Cuetter, A.; Husain, F.; Smith, W.A.; Ekes, R.; et al. Multicentre double-blind study of effect of intrathecally administered natural human fibroblast interferon on exacerbations of multiple sclerosis. Lancet 1986, 2, 1411–1413. [Google Scholar] [CrossRef]
- Panitch, H.S.; Hirsch, R.L.; Haley, A.S.; Johnson, K.P. Exacerbations of multiple sclerosis in patients treated with gamma interferon. Lancet 1987, 1, 893–895. [Google Scholar] [CrossRef]
- Wang, D.; Ghosh, D.; Islam, S.M.; Moorman, C.D.; Thomason, A.E.; Wilkinson, D.S.; Mannie, M.D. IFN-beta facilitates neuroantigen-dependent induction of cd25+ foxp3+ regulatory T cells that suppress experimental autoimmune encephalomyelitis. J. Immunol. 2016, 197, 2992–3007. [Google Scholar] [CrossRef] [PubMed]
- Ramgolam, V.S.; Sha, Y.; Jin, J.; Zhang, X.; Markovic-Plese, S. Ifn-beta inhibits human Th17 cell differentiation. J. Immunol. 2009, 183, 5418–5427. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.P.; Ma, D.H.; Wei, L.; van der Meide, P.H.; Mix, E.; Zhu, J. IFN-beta suppresses experimental autoimmune neuritis in lewis rats by inhibiting the migration of inflammatory cells into peripheral nervous tissue. J. Neurosci. Res. 1999, 56, 123–130. [Google Scholar] [CrossRef]
- Pennell, L.M.; Fish, E.N. Interferon-beta regulates dendritic cell activation and migration in experimental autoimmune encephalomyelitis. Immunology 2017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yuan, S.; Cheng, G.; Guo, B. Type i ifn promotes IL-10 production from T cells to suppress Th17 cells and Th17-associated autoimmune inflammation. PLoS ONE 2011, 6, e28432. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, A.; Miyagaki, T.; DiLillo, D.J.; Matsushita, T.; Horikawa, M.; Kountikov, E.I.; Spolski, R.; Poe, J.C.; Leonard, W.J.; Tedder, T.F. Regulatory b cells control T-cell autoimmunity through IL-21-dependent cognate interactions. Nature 2012, 491, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Streeter, H.B.; Rigden, R.; Martin, K.F.; Scolding, N.J.; Wraith, D.C. Preclinical development and first-in-human study of atx-ms-1467 for immunotherapy of ms. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e93. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.Q.; Wittmer, S.; Dalton, D.K. Failure to suppress the expansion of the activated cd4 T cell population in interferon gamma-deficient mice leads to exacerbation of experimental autoimmune encephalomyelitis. J. Exp. Med. 2000, 192, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Ferber, I.A.; Brocke, S.; Taylor-Edwards, C.; Ridgway, W.; Dinisco, C.; Steinman, L.; Dalton, D.; Fathman, C.G. Mice with a disrupted ifn-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J. Immunol. 1996, 156, 5–7. [Google Scholar] [PubMed]
- Krakowski, M.; Owens, T. Interferon-gamma confers resistance to experimental allergic encephalomyelitis. Eur. J. Immunol. 1996, 26, 1641–1646. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Pavelko, K.; Coffman, R.L. Gamma interferon is critical for resistance to theiler’s virus-induced demyelination. J. Virol. 1995, 69, 7286–7290. [Google Scholar] [PubMed]
- Hsieh, C.S.; Macatonia, S.E.; Tripp, C.S.; Wolf, S.F.; O’Garra, A.; Murphy, K.M. Development of th1 cd4+ T cells through IL-12 produced by listeria-induced macrophages. Science 1993, 260, 547–549. [Google Scholar] [CrossRef] [PubMed]
- Manetti, R.; Parronchi, P.; Giudizi, M.G.; Piccinni, M.P.; Maggi, E.; Trinchieri, G.; Romagnani, S. Natural killer cell stimulatory factor (interleukin 12 [IL-12]) induces T helper type 1 (Th1)-specific immune responses and inhibits the development of IL-4-producing Th cells. J. Exp. Med. 1993, 177, 1199–1204. [Google Scholar] [CrossRef] [PubMed]
- Becher, B.; Durell, B.G.; Noelle, R.J. Experimental autoimmune encephalitis and inflammation in the absence of interleukin-12. J. Clin. Investig. 2002, 110, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Gran, B.; Zhang, G.X.; Yu, S.; Li, J.; Chen, X.H.; Ventura, E.S.; Kamoun, M.; Rostami, A. IL-12p35-deficient mice are susceptible to experimental autoimmune encephalomyelitis: Evidence for redundancy in the IL-12 system in the induction of central nervous system autoimmune demyelination. J. Immunol. 2002, 169, 7104–7110. [Google Scholar] [CrossRef] [PubMed]
- Langrish, C.L.; Chen, Y.; Blumenschein, W.M.; Mattson, J.; Basham, B.; Sedgwick, J.D.; McClanahan, T.; Kastelein, R.A.; Cua, D.J. IL-23 drives a pathogenic t cell population that induces autoimmune inflammation. J. Exp. Med. 2005, 201, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-producing cd4+ effector t cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Kang, H.S.; Kim, B.S. Th17 cells enhance viral persistence and inhibit T cell cytotoxicity in a model of chronic virus infection. J. Exp. Med. 2009, 206, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Mangan, P.R.; Harrington, L.E.; O’Quinn, D.B.; Helms, W.S.; Bullard, D.C.; Elson, C.O.; Hatton, R.D.; Wahl, S.M.; Schoeb, T.R.; Weaver, C.T. Transforming growth factor-beta induces development of the T(h)17 lineage. Nature 2006, 441, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector Th17 and regulatory t cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Nurieva, R.; Yang, X.O.; Martinez, G.; Zhang, Y.; Panopoulos, A.D.; Ma, L.; Schluns, K.; Tian, Q.; Watowich, S.S.; Jetten, A.M.; et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature 2007, 448, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Korn, T.; Bettelli, E.; Gao, W.; Awasthi, A.; Jager, A.; Strom, T.B.; Oukka, M.; Kuchroo, V.K. IL-21 initiates an alternative pathway to induce proinflammatory T(h)17 cells. Nature 2007, 448, 484–487. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Awasthi, A.; Yosef, N.; Quintana, F.J.; Xiao, S.; Peters, A.; Wu, C.; Kleinewietfeld, M.; Kunder, S.; Hafler, D.A.; et al. Induction and molecular signature of pathogenic th17 cells. Nat. Immunol. 2012, 13, 991–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Behi, M.; Ciric, B.; Dai, H.; Yan, Y.; Cullimore, M.; Safavi, F.; Zhang, G.X.; Dittel, B.N.; Rostami, A. The encephalitogenicity of T(h)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine gm-csf. Nat. Immunol. 2011, 12, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Komiyama, Y.; Nakae, S.; Matsuki, T.; Nambu, A.; Ishigame, H.; Kakuta, S.; Sudo, K.; Iwakura, Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J. Immunol. 2006, 177, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Codarri, L.; Gyulveszi, G.; Tosevski, V.; Hesske, L.; Fontana, A.; Magnenat, L.; Suter, T.; Becher, B. Rorgammat drives production of the cytokine gm-csf in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat. Immunol. 2011, 12, 560–567. [Google Scholar] [CrossRef] [PubMed]
- McQualter, J.L.; Darwiche, R.; Ewing, C.; Onuki, M.; Kay, T.W.; Hamilton, J.A.; Reid, H.H.; Bernard, C.C. Granulocyte macrophage colony-stimulating factor: A new putative therapeutic target in multiple sclerosis. J. Exp. Med. 2001, 194, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Carrieri, P.B.; Provitera, V.; De Rosa, T.; Tartaglia, G.; Gorga, F.; Perrella, O. Profile of cerebrospinal fluid and serum cytokines in patients with relapsing-remitting multiple sclerosis: A correlation with clinical activity. Immunopharmacol. Immunotoxicol. 1998, 20, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, F.J.; Khademi, M.; Aram, J.; Ammann, S.; Kockum, I.; Constantinescu, C.; Gran, B.; Piehl, F.; Olsson, T.; Codarri, L.; et al. Multiple sclerosis-associated IL2ra polymorphism controls gm-csf production in human th cells. Nat. Commun. 2014, 5, 5056. [Google Scholar] [CrossRef] [PubMed]
- Rasouli, J.; Ciric, B.; Imitola, J.; Gonnella, P.; Hwang, D.; Mahajan, K.; Mari, E.R.; Safavi, F.; Leist, T.P.; Zhang, G.X.; et al. Expression of gm-csf in t cells is increased in multiple sclerosis and suppressed by ifn-beta therapy. J. Immunol. 2015, 194, 5085–5093. [Google Scholar] [CrossRef] [PubMed]
- Cantrell, M.A.; Anderson, D.; Cerretti, D.P.; Price, V.; McKereghan, K.; Tushinski, R.J.; Mochizuki, D.Y.; Larsen, A.; Grabstein, K.; Gillis, S.; et al. Cloning, sequence, and expression of a human granulocyte/macrophage colony-stimulating factor. Proc. Natl. Acad. Sci. USA 1985, 82, 6250–6254. [Google Scholar] [CrossRef] [PubMed]
- Gough, N.M.; Gough, J.; Metcalf, D.; Kelso, A.; Grail, D.; Nicola, N.A.; Burgess, A.W.; Dunn, A.R. Molecular cloning of cdna encoding a murine haematopoietic growth regulator, granulocyte-macrophage colony stimulating factor. Nature 1984, 309, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Liu, C.H.; Roberts, A.I.; Das, J.; Xu, G.; Ren, G.; Zhang, Y.; Zhang, L.; Yuan, Z.R.; Tan, H.S.; et al. Granulocyte-macrophage colony-stimulating factor (gm-csf) and T-cell responses: What we do and don’t know. Cell Res. 2006, 16, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Sato, N.; Arai, K.; Miyajima, A. Expression cloning of the human IL-3 receptor cdna reveals a shared beta subunit for the human IL-3 and gm-csf receptors. Cell 1991, 66, 1165–1174. [Google Scholar] [CrossRef]
- Quelle, F.W.; Sato, N.; Witthuhn, B.A.; Inhorn, R.C.; Eder, M.; Miyajima, A.; Griffin, J.D.; Ihle, J.N. JAK2 associates with the beta c chain of the receptor for granulocyte-macrophage colony-stimulating factor, and its activation requires the membrane-proximal region. Mol. Cell. Biol. 1994, 14, 4335–4341. [Google Scholar] [CrossRef] [PubMed]
- Croxford, A.L.; Lanzinger, M.; Hartmann, F.J.; Schreiner, B.; Mair, F.; Pelczar, P.; Clausen, B.E.; Jung, S.; Greter, M.; Becher, B. The cytokine gm-csf drives the inflammatory signature of ccr2+ monocytes and licenses autoimmunity. Immunity 2015, 43, 502–514. [Google Scholar] [CrossRef] [PubMed]
- Van de Laar, L.; Coffer, P.J.; Woltman, A.M. Regulation of dendritic cell development by gm-csf: Molecular control and implications for immune homeostasis and therapy. Blood 2012, 119, 3383–3393. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.D.; Mueller, M.; Chou, T.H. Role of granulocyte/macrophage colony-stimulating factor in the regulation of murine alveolar macrophage proliferation and differentiation. J. Immunol. 1988, 141, 139–144. [Google Scholar] [PubMed]
- Shibata, Y.; Berclaz, P.Y.; Chroneos, Z.C.; Yoshida, M.; Whitsett, J.A.; Trapnell, B.C. Gm-csf regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity 2001, 15, 557–567. [Google Scholar] [CrossRef]
- Suzuki, T.; Sakagami, T.; Rubin, B.K.; Nogee, L.M.; Wood, R.E.; Zimmerman, S.L.; Smolarek, T.; Dishop, M.K.; Wert, S.E.; Whitsett, J.A.; et al. Familial pulmonary alveolar proteinosis caused by mutations in csf2ra. J. Exp. Med. 2008, 205, 2703–2710. [Google Scholar] [CrossRef] [PubMed]
- Dirksen, U.; Nishinakamura, R.; Groneck, P.; Hattenhorst, U.; Nogee, L.; Murray, R.; Burdach, S. Human pulmonary alveolar proteinosis associated with a defect in gm-csf/IL-3/IL-5 receptor common beta chain expression. J. Clin. Investig. 1997, 100, 2211–2217. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Tanaka, N.; Watanabe, J.; Uchida; Kanegasaki, S.; Yamada, Y.; Nakata, K. Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 1999, 190, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Inaba, K.; Inaba, M.; Romani, N.; Aya, H.; Deguchi, M.; Ikehara, S.; Muramatsu, S.; Steinman, R.M. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 1992, 176, 1693–1702. [Google Scholar] [CrossRef] [PubMed]
- Inaba, K.; Inaba, M.; Deguchi, M.; Hagi, K.; Yasumizu, R.; Ikehara, S.; Muramatsu, S.; Steinman, R.M. Granulocytes, macrophages, and dendritic cells arise from a common major histocompatibility complex class ii-negative progenitor in mouse bone marrow. Proc. Natl. Acad. Sci. USA 1993, 90, 3038–3042. [Google Scholar] [CrossRef] [PubMed]
- Helft, J.; Bottcher, J.; Chakravarty, P.; Zelenay, S.; Huotari, J.; Schraml, B.U.; Goubau, D.; Reis e Sousa, C. Gm-csf mouse bone marrow cultures comprise a heterogeneous population of cd11c(+)mhcii(+) macrophages and dendritic cells. Immunity 2015, 42, 1197–1211. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.J.; Brady, J.L.; Ryg-Cornejo, V.; Hansen, D.S.; Vremec, D.; Shortman, K.; Zhan, Y.; Lew, A.M. Gm-csf-responsive monocyte-derived dendritic cells are pivotal in th17 pathogenesis. J. Immunol. 2014, 192, 2202–2209. [Google Scholar] [CrossRef] [PubMed]
- Spath, S.; Komuczki, J.; Hermann, M.; Pelczar, P.; Mair, F.; Schreiner, B.; Becher, B. Dysregulation of the cytokine gm-csf induces spontaneous phagocyte invasion and immunopathology in the central nervous system. Immunity 2017, 46, 245–260. [Google Scholar] [CrossRef] [PubMed]
- Mufazalov, I.A.; Schelmbauer, C.; Regen, T.; Kuschmann, J.; Wanke, F.; Gabriel, L.A.; Hauptmann, J.; Muller, W.; Pinteaux, E.; Kurschus, F.C.; et al. IL-1 signaling is critical for expansion but not generation of autoreactive gm-csf+ th17 cells. EMBO J. 2017, 36, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Ulges, A.; Witsch, E.J.; Pramanik, G.; Klein, M.; Birkner, K.; Buhler, U.; Wasser, B.; Luessi, F.; Stergiou, N.; Dietzen, S.; et al. Protein kinase ck2 governs the molecular decision between encephalitogenic Th17 cell and treg cell development. Proc. Natl. Acad. Sci. USA 2016, 113, 10145–10150. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.; Yang, F.; Zhou, Y.; Yang, H.; Low, P.Y.; Kemeny, D.M.; Tan, P.; Moh, A.; Kaplan, M.H.; Zhang, Y.; et al. Stat5 programs a distinct subset of gm-csf-producing t helper cells that is essential for autoimmune neuroinflammation. Cell Res. 2014, 24, 1387–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukens, J.R.; Barr, M.J.; Chaplin, D.D.; Chi, H.; Kanneganti, T.D. Inflammasome-derived IL-1beta regulates the production of gm-csf by cd4(+) t cells and gammadelta T cells. J. Immunol. 2012, 188, 3107–3115. [Google Scholar] [CrossRef] [PubMed]
- Noster, R.; Riedel, R.; Mashreghi, M.F.; Radbruch, H.; Harms, L.; Haftmann, C.; Chang, H.D.; Radbruch, A.; Zielinski, C.E. IL-17 and gm-csf expression are antagonistically regulated by human T helper cells. Sci. Transl. Med. 2014, 6, 241ra280. [Google Scholar] [CrossRef] [PubMed]
- Segal, B.M.; Constantinescu, C.S.; Raychaudhuri, A.; Kim, L.; Fidelus-Gort, R.; Kasper, L.H.; Ustekinumab, M.S.I. Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: A phase II, double-blind, placebo-controlled, randomised, dose-ranging study. Lancet Neurol. 2008, 7, 796–804. [Google Scholar] [CrossRef]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Rezk, A.; Miyazaki, Y.; Hilgenberg, E.; Touil, H.; Shen, P.; Moore, C.S.; Michel, L.; Althekair, F.; Rajasekharan, S.; et al. Proinflammatory gm-csf-producing b cells in multiple sclerosis and B cell depletion therapy. Sci. Transl. Med. 2015, 7, 310ra166. [Google Scholar] [CrossRef] [PubMed]
- Ifergan, I.; Davidson, T.S.; Kebir, H.; Xu, D.; Palacios-Macapagal, D.; Cann, J.; Rodgers, J.M.; Hunter, Z.N.; Pittet, C.L.; Beddow, S.; et al. Targeting the GM-CSF receptor for the treatment of CNS autoimmunity. J. Autoimmun. 2017. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, C.S.; Asher, A.; Fryze, W.; Kozubski, W.; Wagner, F.; Aram, J.; Tanasescu, R.; Korolkiewicz, R.P.; Dirnberger-Hertweck, M.; Steidl, S.; et al. Randomized phase 1b trial of mor103, a human antibody to gm-csf, in multiple sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e117. [Google Scholar] [CrossRef] [PubMed]
- Teige, I.; Treschow, A.; Teige, A.; Mattsson, R.; Navikas, V.; Leanderson, T.; Holmdahl, R.; Issazadeh-Navikas, S. IFN-beta gene deletion leads to augmented and chronic demyelinating experimental autoimmune encephalomyelitis. J. Immunol. 2003, 170, 4776–4784. [Google Scholar] [CrossRef] [PubMed]
- Limmroth, V.; Putzki, N.; Kachuck, N.J. The interferon beta therapies for treatment of relapsing-remitting multiple sclerosis: Are they equally efficacious? A comparative review of open-label studies evaluating the efficacy, safety, or dosing of different interferon beta formulations alone or in combination. Ther. Adv. Neurol. Disord. 2011, 4, 281–296. [Google Scholar] [PubMed]
- Sutton, C.; Brereton, C.; Keogh, B.; Mills, K.H.; Lavelle, E.C. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J. Exp. Med. 2006, 203, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Prabhu Das, M.; Howard, E.D.; Weiner, H.L.; Sobel, R.A.; Kuchroo, V.K. IL-10 is critical in the regulation of autoimmune encephalomyelitis as demonstrated by studies of IL-10- and IL-4-deficient and transgenic mice. J. Immunol. 1998, 161, 3299–3306. [Google Scholar] [PubMed]
- Havrdova, E.; Belova, A.; Goloborodko, A.; Tisserant, A.; Wright, A.; Wallstroem, E.; Garren, H.; Maguire, R.P.; Johns, D.R. Activity of secukinumab, an anti-IL-17a antibody, on brain lesions in rrms: Results from a randomized, proof-of-concept study. J. Neurol. 2016, 263, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, R.D.; Zehntner, S.P.; Kelly, L.M.; Bourbonniere, L.; Owens, T. Elevated interferon gamma expression in the central nervous system of tumour necrosis factor receptor 1-deficient mice with experimental autoimmune encephalomyelitis. Immunology 2006, 118, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Titelbaum, D.S.; Degenhardt, A.; Kinkel, R.P. Anti-tumor necrosis factor alpha-associated multiple sclerosis. Am. J. Neuroradiol. 2005, 26, 1548–1550. [Google Scholar] [PubMed]


{kind=link}
{kind=link}
| Cytokine | Main Producers | Levels in MS Patients | Role in EAE | Potential Treatments of MS |
|---|---|---|---|---|
| GM-CSF | T cells | Elevated | GM-CSF-deficient mice are completely resistant to EAE [123] | Phase 1b trial of humanized anti-GM-CSF mAb MOR103 in MS is completed [153] |
| IFN-β | pDCs | Not reported | Ifnb−/− mice exhibit increased EAE severity [154] | First line treatment of RRMS [155] |
| IFN-γ | Th1 cells, NK cells, NKT cells | Elevated | Ifng−/− mice exhibit increased EAE severity [105] | Intravenous infusion of IFN-γ exacerbates disease in MS patients [96] |
| IL-1β | Monocytes, macrophages | Elevated | Il1r1−/− mice are resistant to EAE [156] | Not reported |
| IL-10 | Tregs, macrophages, DCs, B cells | Reduced | Il10−/− mice exhibit increased EAE severity [157] | Not reported |
| IL-12 | DCs, macrophages | Elevated | IL-12 p35−/− exhibit increased EAE severity [110] | Anti-IL-12/IL-23 p40 mAb Ustekinumab does not show efficacy in treating RRMS in phase II trial [149] |
| IL-17 | Th17 cells, γδ T cells, NKT cells | Elevated | Il17a−/− mice are partially resistant to EAE [121] | Anti-17A mAb Secukinumab reduces disease severity in RRMS patients [158] |
| IL-23 | DCs, macrophages | Elevated | Il23r−/− mice are completely resistant to EAE [47] | Anti-IL-12/IL-23 p40 mAb Ustekinumab does not show efficacy in treating RRMS in phase II trial [149] |
| TNF-α | Macrophages | Elevated | Tnfrsf1a−/− mice are partially resistant to EAE [159] | Treatment of MS patients with anti-TNF-α exacerbates disease in MS patients [160] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palle, P.; Monaghan, K.L.; Milne, S.M.; Wan, E.C.K. Cytokine Signaling in Multiple Sclerosis and Its Therapeutic Applications. Med. Sci. 2017, 5, 23. https://doi.org/10.3390/medsci5040023
Palle P, Monaghan KL, Milne SM, Wan ECK. Cytokine Signaling in Multiple Sclerosis and Its Therapeutic Applications. Medical Sciences. 2017; 5(4):23. https://doi.org/10.3390/medsci5040023
Chicago/Turabian StylePalle, Pushpalatha, Kelly L. Monaghan, Sarah M. Milne, and Edwin C.K. Wan. 2017. "Cytokine Signaling in Multiple Sclerosis and Its Therapeutic Applications" Medical Sciences 5, no. 4: 23. https://doi.org/10.3390/medsci5040023
APA StylePalle, P., Monaghan, K. L., Milne, S. M., & Wan, E. C. K. (2017). Cytokine Signaling in Multiple Sclerosis and Its Therapeutic Applications. Medical Sciences, 5(4), 23. https://doi.org/10.3390/medsci5040023