Results of Beta Secretase-Inhibitor Clinical Trials Support Amyloid Precursor Protein-Independent Generation of Beta Amyloid in Sporadic Alzheimer’s Disease

Abstract
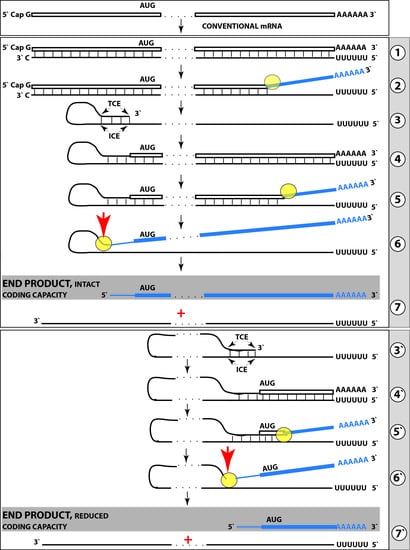
:
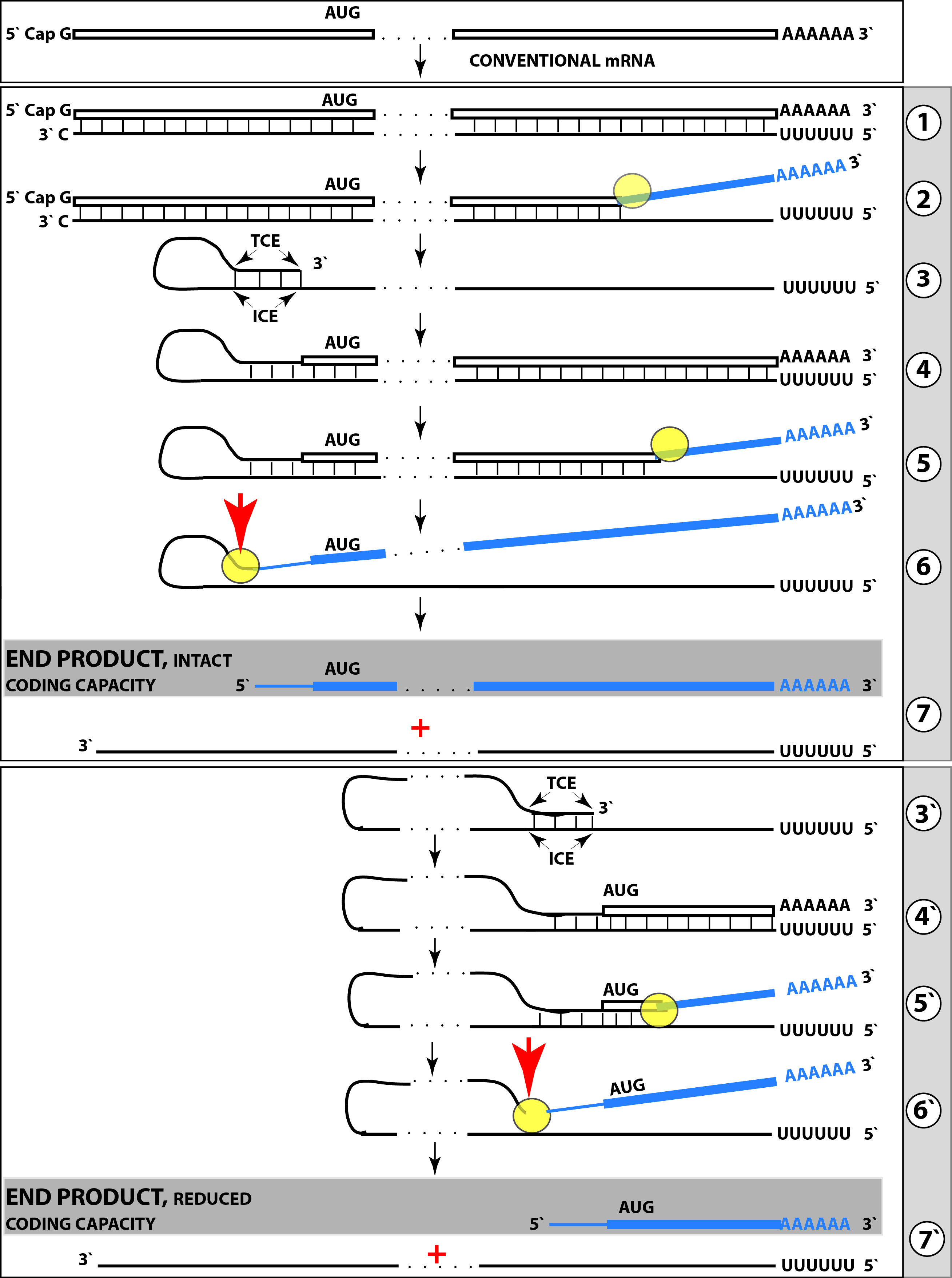{kind=link}
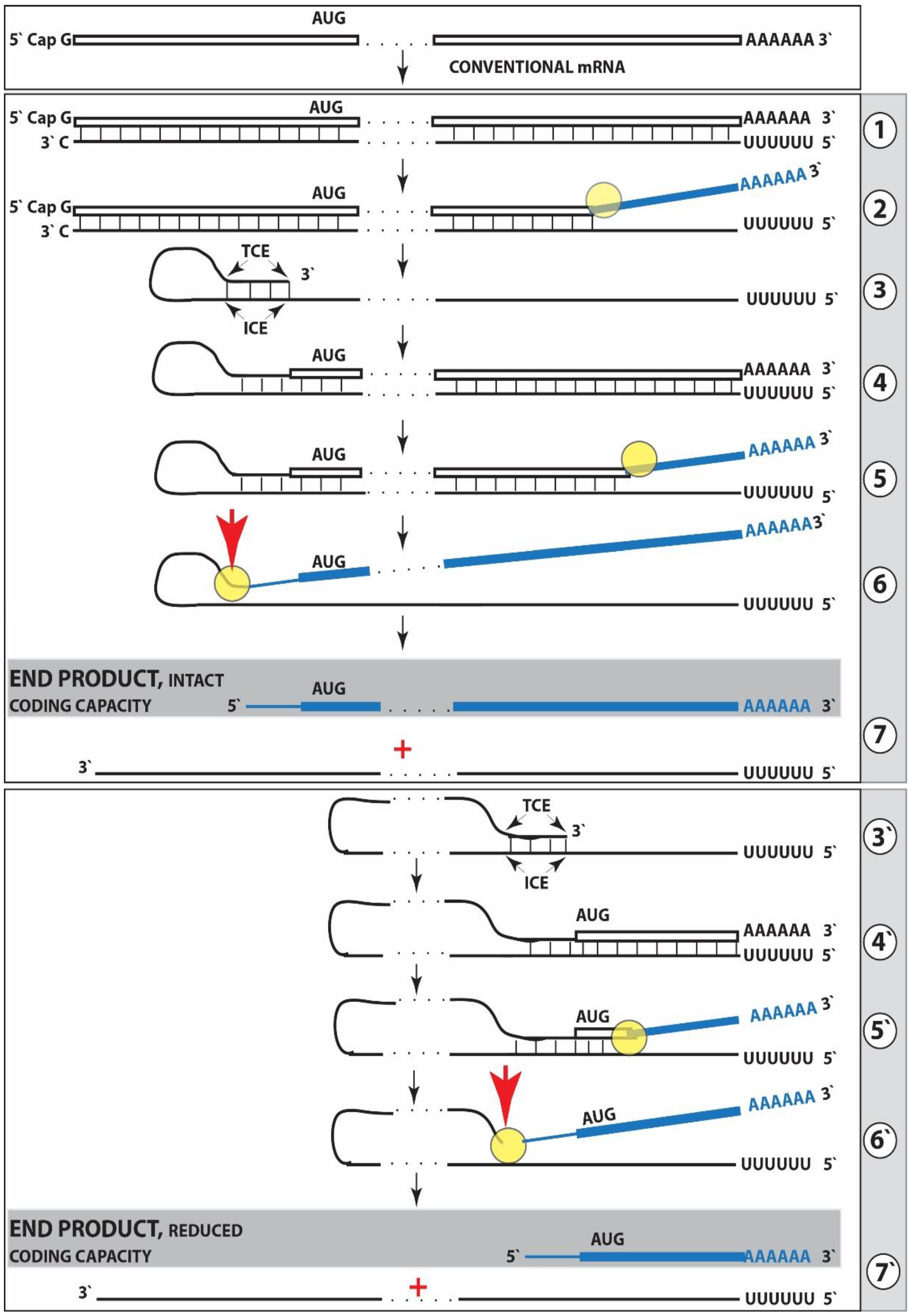{kind=link}
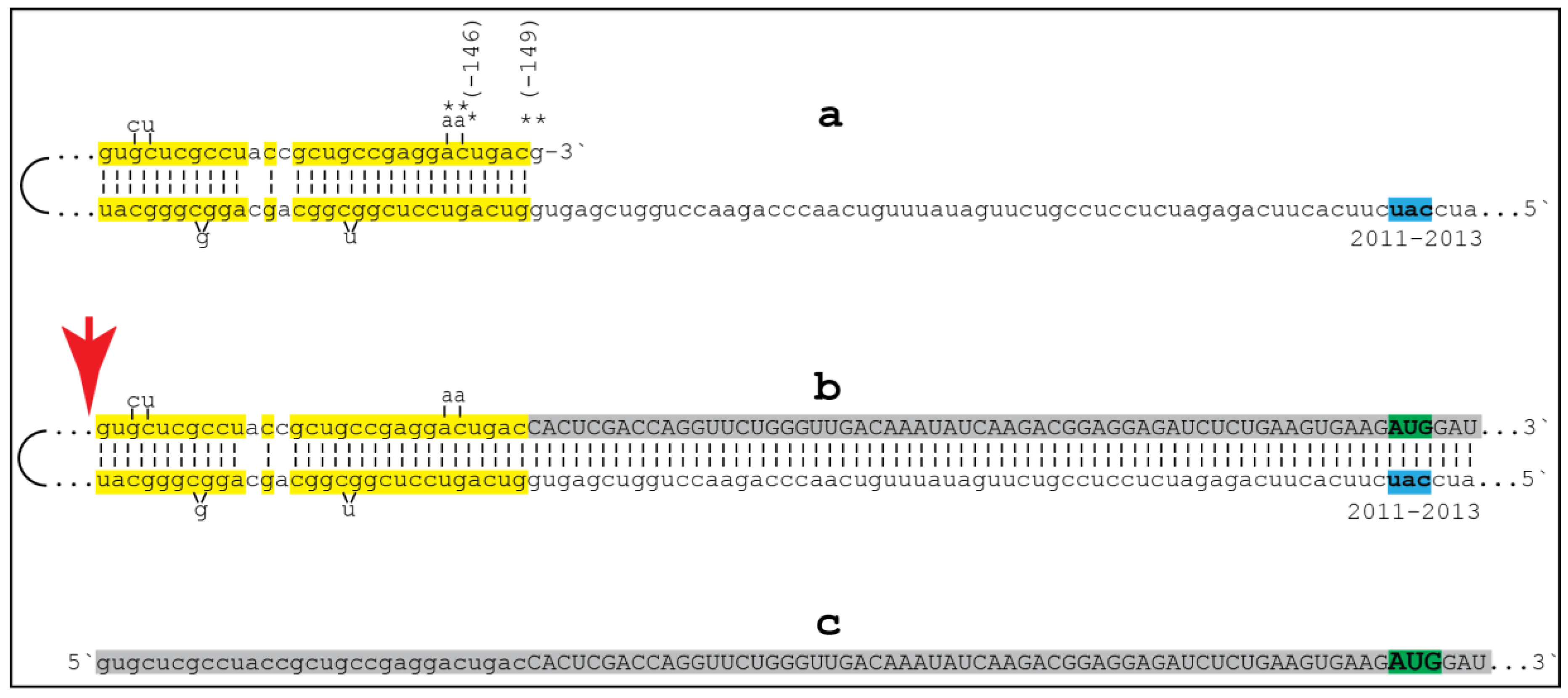{kind=link}
1. Introduction
2. Success of β-site Amyloid Precursor Protein-Cleaving Enzyme Inhibitors in Preclinical Tests
3. Inhibition of Beta Secretase Activity Rescues Functional Impairments in Animal Models of Familial Alzheimer’s Disease
4. BACE Inhibitors Are Completely Inefficient in Treatment of Sporadic Alzheimer’s Disease
5. Results of Clinical Trials Can Be Explained by APP-Independent and BACE Inhibition-Insensitive Generation of Beta Amyloid in Sporadic Alzheimer’s Disease
6. Generation of Severely 5′-Truncated βAPP mRNA Encoding C99 Fragment of β Amyloid Precursor Protein in Sporadic Alzheimer’s Disease
7. Results of Clinical Trials Confirm Key Predictions for Amyloid Precursor Protein-Independent Generation of Beta Amyloid in Sporadic Alzheimer’s Disease
8. BACE Inhibitors Deemed Failed in Sporadic AD Trials Could Be Perfectly Suitable for Treatment of Familial AD
9. Lessons from the Trials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Haass, C.; Lemere, C.; Capell, A.; Citron, M.; Seubert, P.; Schenk, D.; Lannfelt, L.; Selkoe, D. The Swedish mutation causes early-onset Alzheimer’s disease by beta-secretase cleavage within the secretory pathway. Nat. Med. 1995, 1, 1291–1296. [Google Scholar] [CrossRef] [PubMed]
- Dyrks, T.; Dyrks, E.; Monning, U.; Urmoneit, B.; Turner, J.; Beyreuther, K. Generation of βA4 from the amyloid protein precursor and fragments thereof. FEBS Lett. 1993, 335, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Iizuka, T.; Shoji, M.; Kawarabayashi, T.; Sato, M.; Kobayashi, T.; Tada, N.; Kasai, K.; Matsubara, E.; Watanabe, M.; Tomidokoro, Y.; et al. Intracellular generation of amyloid β-protein from amyloid β-protein precursor fragment by direct cleavage with β- and γ-secretase. Biochem. Biophys. Res. Commun. 1996, 218, 238–242. [Google Scholar] [CrossRef] [PubMed]
- DeStrooper, B.; Annaert, W. Proteolytic processing and cell biological functions of the amyloid precursor protein. J. Cell Sci. 2000, 113, 1857–1870. [Google Scholar]
- Barber, R. The genetics of Alzheimer’s disease. Scientifica 2012, 2012, 246210. [Google Scholar] [CrossRef] [PubMed]
- Vassar, R. BACE1 inhibitors drugs in clinical trials for Alzheimer’s disease. Alzheimer’s Res. Ther. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Beyreuther, K.; Masters, C. Amyloid precursor protein (APP) and BZA4 amyloid in the etiology of Alzheimer’s disease: Precursor-product relationships in the derangement of neuronal function. Brain Pathol. 1991, 1, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Selkoe, D. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef]
- Hardy, J.; Higgins, G. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R. Mitochondria and mitochondrial cascades in Alzheimer’s disease. J. Alzheimer's Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.; Brayne, C. Understanding the roles of mutations in the amyloid precursor protein in Alzheimer disease. Mol. Psych. 2018, 23, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, Z.; Cai, F.; Wu, Y.; Zhang, J.; Song, W. BACE1 Cleavage Site Selection Critical for Amyloidogenesis and Alzheimer’s Pathogenesis. J. Neurosci. 2017, 37, 6915–6925. [Google Scholar] [CrossRef] [PubMed]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Hussain, I.; Powell, D.; Howlett, D.R.; Tew, D.G.; Meek, T.D.; Chapman, C.; Gloger, I.S.; Murphy, K.E.; Southan, C.D.; Ryan, D.M.; et al. Identification of novel aspartic protease (Asp2) as beta secretase. Mol. Cell. Neurosci. 1999, 14, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Anderson, J.; Barbour, R.; Basl, G.; Caccavello, R.; Davis, D.; Doan, M.; Dovey, H.; Frigon, N.; Hong, J.; et al. Purification and cloning of amyloid precursor protein β-secretase from human brain. Nature 1999, 402, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Strickland, C.; Voigt, J.; Kennedy, M.; Beyer, B.; Senior, M.; Smith, E.; Nechuta, T.; Madison, V.; Czarniecki, M.; et al. Application of fragment-based NMR screening, X-ray crystallography, structure-based design, and focused chemical library design to identify novel μM leads for the development of nM BACE-1 inhibitors. J. Med. Chem. 2010, 53, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Sun, Z.; Ye, Y.; Voigt, J.; Strickland, C.; Smith, E.; Cumming, J.; Wang, L.; Wong, J.; Wang, Y.; et al. Discovery of cyclic acylguanidines as highly potent and selective β-site amyloid cleaving enzyme (BACE) inhibitors: Part I—Inhibitor design and validation. J. Med. Chem. 2010, 53, 951–965. [Google Scholar] [CrossRef] [PubMed]
- Cumming, J.; Smith, E.; Wang, L.; Misiaszek, J.; Durkin, J.; Pan, J.; Iserloh, U.; Wu, Y.; Zhu, Z.; Strickland, C.; et al. Structure based design of iminohydantoin BACE1 inhibitors: Identification of an orally available, centrally active BACE1 inhibitor. Bioorg. Med. Chem. Lett. 2012, 22, 2444–2449. [Google Scholar] [CrossRef] [PubMed]
- Edwards, P.; Albert, J.; Sylvester, M.; Aharony, D.; Andisik, D.; Callaghan, O.; Campbell, J.; Carr, R.; Chessari, G.; Congreve, M.; et al. Application of fragment-based lead generation to the discovery of novel, cyclic amidine β-secretase inhibitors with nanomolar potency, cellular activity, and high ligand efficiency. J. Med. Chem. 2007, 50, 5912–5925. [Google Scholar] [CrossRef] [PubMed]
- Barrow, J.; Stauffer, S.; Rittle, K.; Ngo, P.; Yang, Z.; Selnick, H.; Graham, S.; Munshi, S.; McGaughey, G.; Holloway, M.; et al. Discovery and X-ray crystallographic analysis of a S-piropiperidine iminohydantoin inhibitor of β-secretase. J. Med. Chem. 2008, 51, 6259–6262. [Google Scholar] [CrossRef] [PubMed]
- Malamas, M.; Erdei, J.; Gunawan, I.; Turner, J.; Hu, Y.; Wagner, E.; Fan, K.; Chopra, R.; Olland, A.; Bard, J.; et al. Design and synthesis of 5,5′-disubstituted aminohydantoins as potent and selective human β-secretase (BACE1) inhibitors. J. Med. Chem. 2010, 53, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Rueeger, H.; Rondeau, J.; McCarthy, C.; Moebitz, H.; Tintelnot-Blomley, M.; Neumann, U.; Desrayaud, S. Structure based design, synthesis and SAR of cyclic hydroxyethylamine (HEA) BACE-1 inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 1942–1947. [Google Scholar] [CrossRef] [PubMed]
- Probst, G.; Xu, Y. Small-Molecule BACE1 Inhibitors: A patent literature review (2006−2011). Expert Opin. Ther. Patents 2012, 22, 511–540. [Google Scholar] [CrossRef] [PubMed]
- May, P.; Dean, R.; Lowe, S.; Martenyi, F.; Sheehan, S.; Boggs, L.; Monk, S.; Mathes, B.; Mergott, D.; Watson, B.; et al. Robust central reduction of amyloid-β in humans with an orally available, non-peptidic β-secretase inhibitor. J. Neurosci. 2011, 31, 6507–6516. [Google Scholar] [CrossRef] [PubMed]
- Stamford, A.W.; Scott, J.D.; Li, S.W.; Babu, S.; Tadesse, D.; Hunter, R.; Wu, Y.; Misiaszek, J.; Cumming, J.N.; Gilbert, E.J.; et al. Discovery of an orally available, brain penetrant BACE1 inhibitor that affords robust CNS Aβ reduction. ACS Med. Chem. Lett. 2012, 3, 897–902. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.E.; Stamford, A.W.; Chen, X.; Cox, K.; Cumming, J.N.; Dockendorf, M.F.; Egan, M.; Ereshefsky, M.; Hodgson, R.A.; Hyde, L.A.; et al. The BACE1 inhibitor verubecestat (MK-8931) reduces CNS b-amyloid in animal models and in Alzheimer’s disease patients. Sci. Transl. Med. 2016, 8, 363ra150. [Google Scholar] [CrossRef] [PubMed]
- Keskin, A.D.; Kekuš, M.; Adelsberger, H.; Neumann, U.; Shimshek, D.R.; Song, B.; Zott, B.; Peng, T.; Förstl, H.; Staufenbiel, M.; et al. BACE inhibition-dependent repair of Alzheimer’s pathophysiology. Proc. Natl. Acad. Sci. USA 2017, 114, 8631–8636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, U.; Rueeger, H.; Machauer, R.; Veenstra, S.J.; Lueoend, R.M.; Tintelnot-Blomley, M.; Laue, G.; Beltz, K.; Vogg, B.; Schmid, P.; et al. A novel BACE inhibitor NB-360 shows a superior pharmacological profile and robust reduction of amyloid-β and neuroinflammation in APP transgenic mice. Mol. Neurodegen. 2015, 10, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Das, B.; Hou, H.; He, W.; Yan, R. BACE1 deletion in the adult mouse reverses preformed amyloid deposition and improves cognitive functions. J. Exp. Med. 2018, 215, 927–940. [Google Scholar] [CrossRef] [PubMed]
- Nativio, R.; Donahue, G.; Berson, A.; Lan, Y.; Amlie-Wolf, A.; Tuzer, F.; Toledo, J.B.; Gosai, S.J.; Gregory, B.D.; Torres, C.; et al. Dysregulation of the epigenetic landscape of normal aging in Alzheimer’s disease. Nat. Neurosci. 2018, 21, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Volloch, V. A mechanism for ß-amyloid overproduction in Alzheimer’s disease: Precursor-independent generation of ß-amyloid via antisense RNA-primed mRNA synthesis. FEBS Lett. 1996, 390, 124–128. [Google Scholar] [CrossRef]
- Volloch, V. Mechanism for β-amyloid overproduction in Alzheimer’s Disease: Possible antisense RNA-mediated generation of a 5’-truncated βAPP mRNA encoding 12 kDa C-terminal fragment of βAPP, the immediate precursor of Aß. In Molecular Mechanisms of Dementia; Wasco, W., Tanzi, R., Eds.; Humana Press: Totowa, NJ, USA, 1997. [Google Scholar]
- Volloch, V. Possible mechanism for resistance to Alzheimer's disease (AD) in mice suggests new approach to generate a mouse model for sporadic AD and may explain familial resistance to AD in man. Exp. Neurobiol. 1997, 144, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Breimer, L.; Denny, P. Alzheimer amyloid aspects. Nature 1987, 326, 749–750. [Google Scholar] [CrossRef] [PubMed]
- Citron, M.; Haass, C.; Selkoe, D. Production of amyloid-β-peptide by cultured cells: No evidence for internal initiation of translation at Met596. Neurobiol. Aging 1993, 14, 571–573. [Google Scholar] [CrossRef]
- Volloch, V.; Schweitzer, B.; Rits, S. Antisense globin RNA in mouse erythroid tissues: Structure, origin, and possible function. Proc. Natl. Acad. Sci. USA 1996, 93, 2476–2481. [Google Scholar] [CrossRef] [PubMed]
- Rits, S.; Olsen, B.; Volloch, V. RNA-dependent synthesis of mammalian mRNA: Identification of chimeric intermediate and putative end-product. BioRxiv 2016. [Google Scholar] [CrossRef]
- Kapranov, P.; Ozsolak, F.; Kim, S.; Foissac, S.; Lipson, D.; Hart, C.; Roels, S.; Borel, C.; Antonarakis, S.; Monaghan, A.; et al. New class of gene-termini-associated human RNAs suggests a novel RNA copying mechanism. Nature 2010, 466, 642–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mita, S.; Sadlock, J.; Herbert, J.; Schon, E. A cDNA specifying the human amyloid beta precursor protein encodes a 95-kDa polypeptide. Nucl. Acids Res. 1988, 16, 9351. [Google Scholar] [CrossRef] [PubMed]
- Mita, S.; Sadlock, J.; Herbert, J.; Schon, E. A cDNA specifying the human amyloid beta precursor protein encodes a 95-kDa polypeptide: CORRECTION. Nucl. Acids Res. 1988, 16, 11402. [Google Scholar] [CrossRef]
- Salbaum, J.; Weidemann, A.; Lemaire, H.; Masters, C.; Beyreuther, K. The promoter of Alzheimer’s disease A4 precursor gene. EMBO J. 1988, 7, 2807–2813. [Google Scholar] [PubMed]
- Hebert, S.; Horre, K.; Nicolaï, L.; Papadopoulou, A.; Mandemakers, W.; Silahtaroglu, A.; Kauppinen, S.; Delacourte, A.; De Strooper, B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc. Natl. Acad. Sci. USA 2008, 105, 6415–6420. [Google Scholar] [CrossRef] [PubMed]
- Zanchez-Mut, J.V.; Heyn, H.; Silva, B.A.; Dixsaut, L.; Garcia-Esparcia, P.; Vidal, E.; Sayols, S.; Glauser, L.; Monteagudo-Sánchez, A.; Perez-Tur, J.; et al. PM20D1 is a quantitative trait locus associated with Alzheimer’s disease. Nat. Med. 2018, 24, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.; Sagare, A.; Zlokovic, B. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Volloch, V.; Rits, S. Results of Beta Secretase-Inhibitor Clinical Trials Support Amyloid Precursor Protein-Independent Generation of Beta Amyloid in Sporadic Alzheimer’s Disease. Med. Sci. 2018, 6, 45. https://doi.org/10.3390/medsci6020045
Volloch V, Rits S. Results of Beta Secretase-Inhibitor Clinical Trials Support Amyloid Precursor Protein-Independent Generation of Beta Amyloid in Sporadic Alzheimer’s Disease. Medical Sciences. 2018; 6(2):45. https://doi.org/10.3390/medsci6020045
Chicago/Turabian StyleVolloch, Vladimir, and Sophia Rits. 2018. "Results of Beta Secretase-Inhibitor Clinical Trials Support Amyloid Precursor Protein-Independent Generation of Beta Amyloid in Sporadic Alzheimer’s Disease" Medical Sciences 6, no. 2: 45. https://doi.org/10.3390/medsci6020045
APA StyleVolloch, V., & Rits, S. (2018). Results of Beta Secretase-Inhibitor Clinical Trials Support Amyloid Precursor Protein-Independent Generation of Beta Amyloid in Sporadic Alzheimer’s Disease. Medical Sciences, 6(2), 45. https://doi.org/10.3390/medsci6020045