Substitution Effects on the Optoelectronic Properties of Coumarin Derivatives

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Computational Method
2.1. Molecule Preparation and DFT/TD-DFT Calculations
2.2. Molecular Dynamics (MD) Simulations
3. Results and Discussion
3.1. Electronic Properties of the Compounds Using B3LYP XC Functional
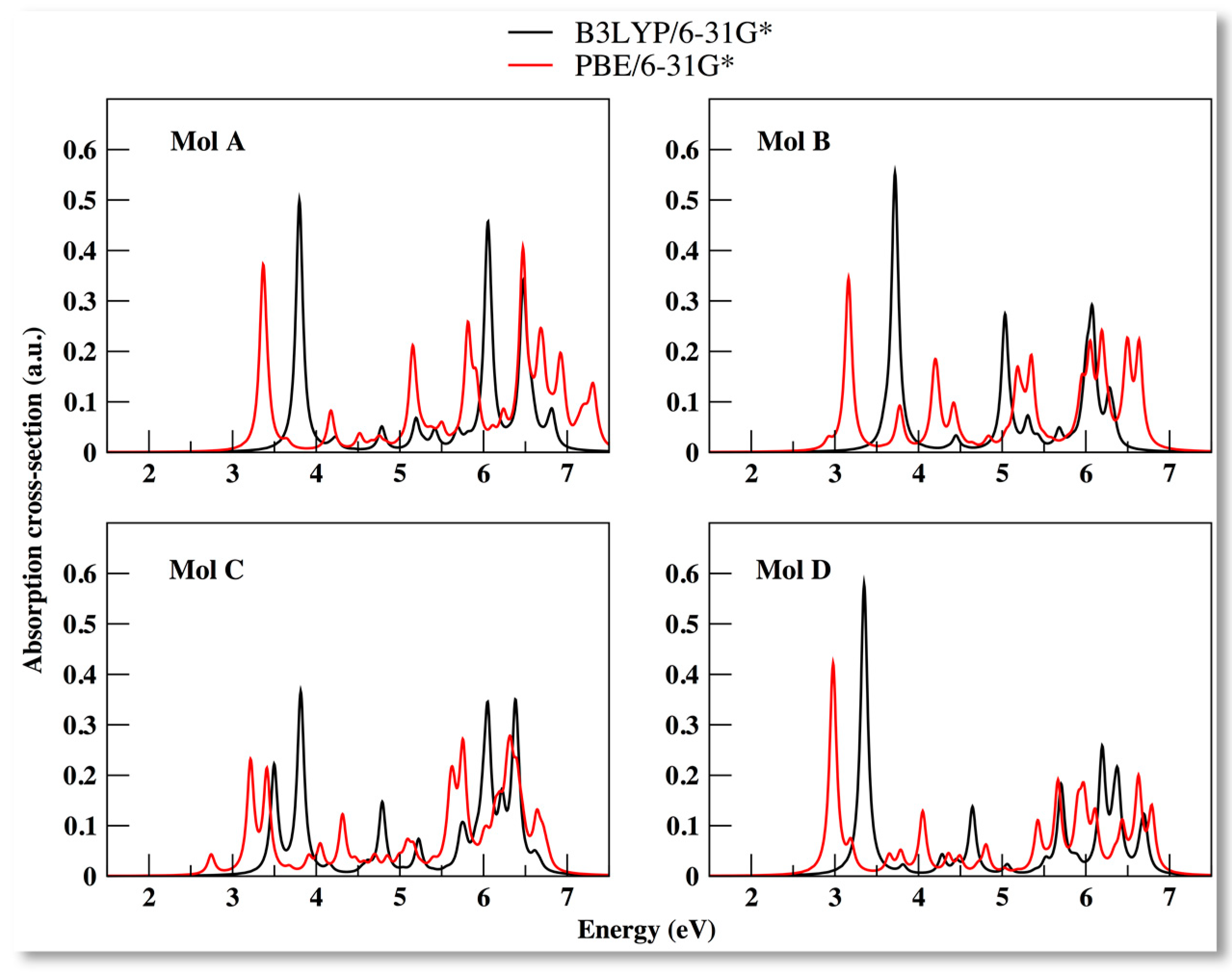
3.2. Electronic Properties of the Compounds Using PBE XC Functional
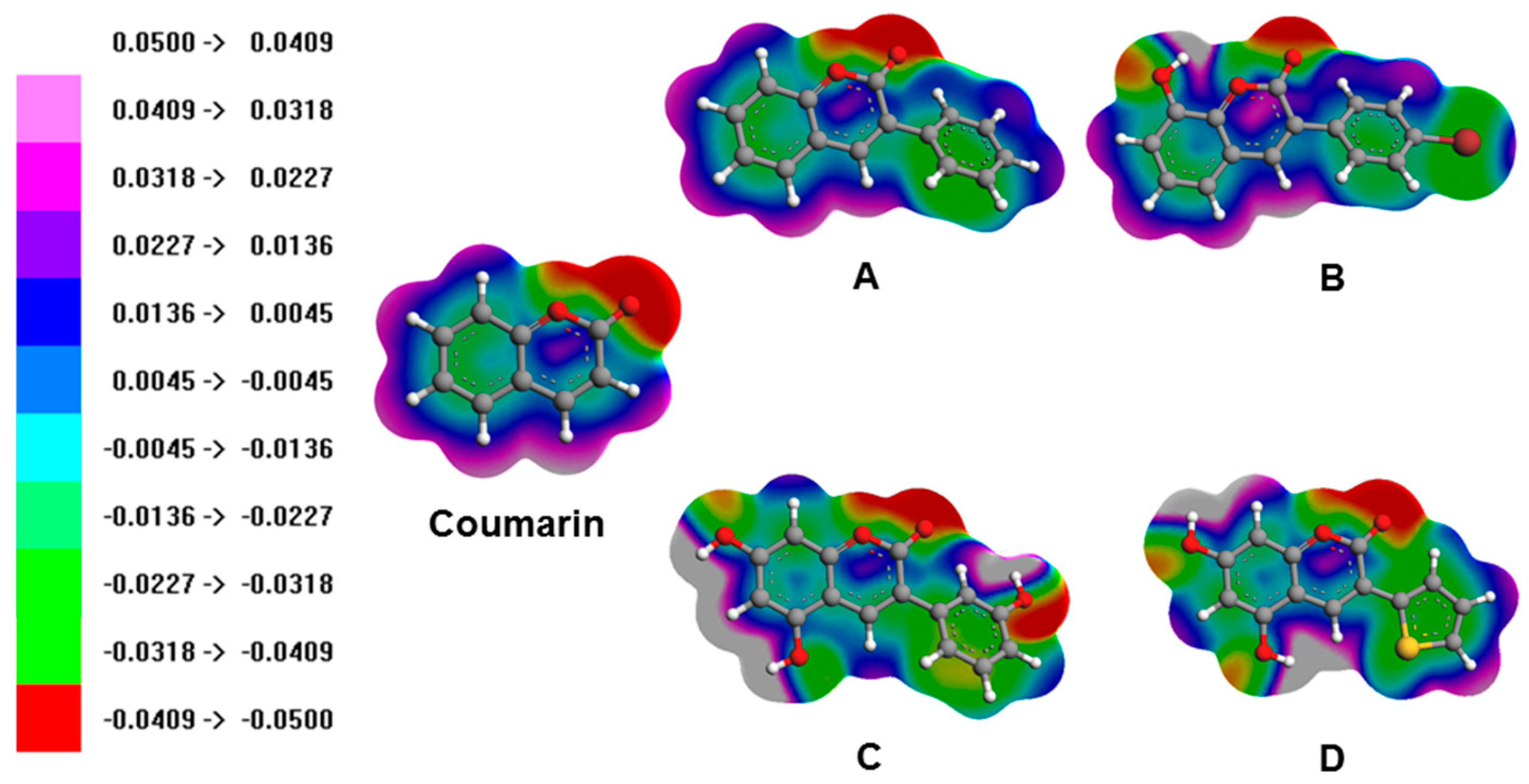
3.3. Molecular Orbitals and Electrostatic Potentials
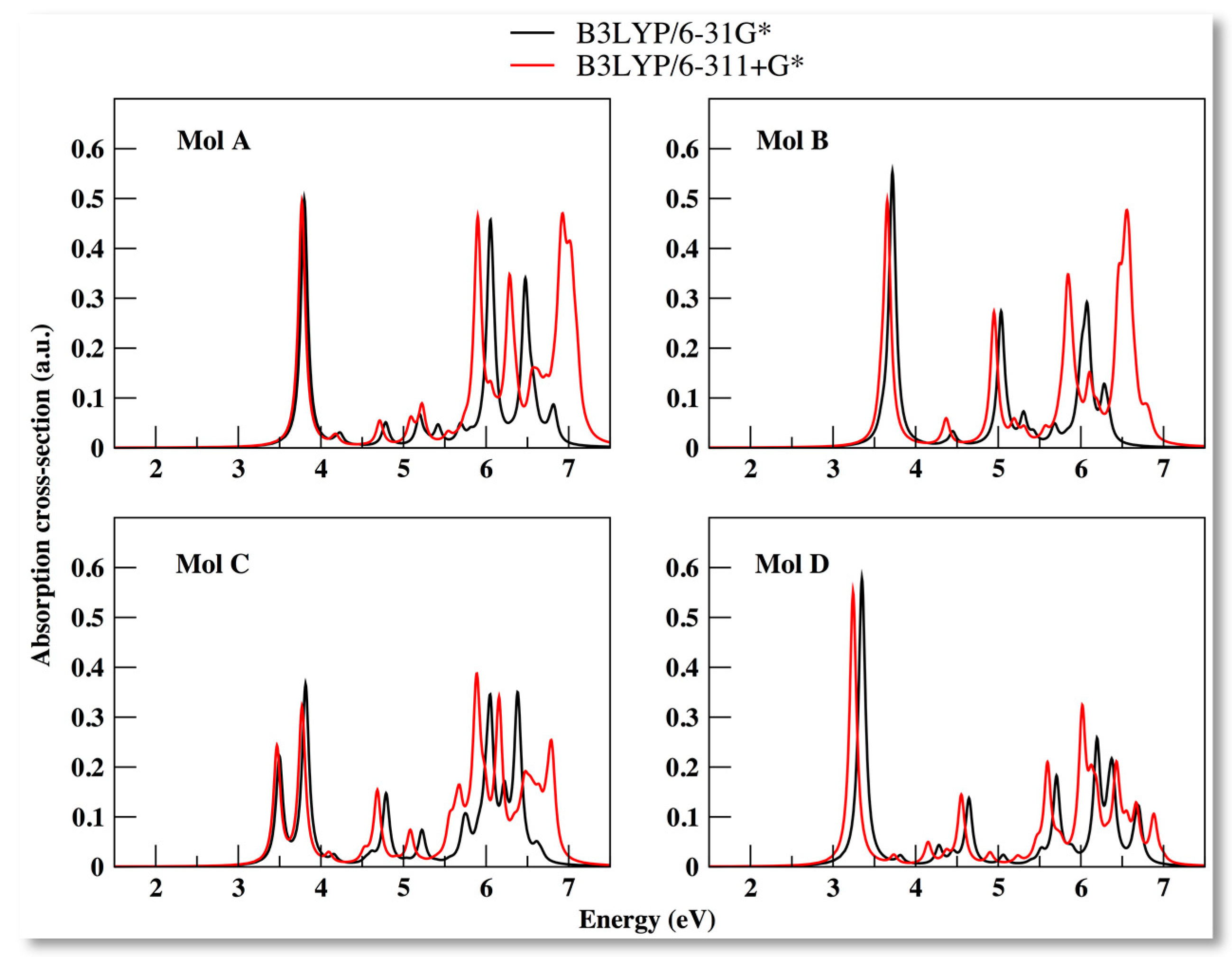
3.4. Optical Properties
3.5. Solvent Effects on the Absorption Spectra for the Coumarin Derivatives
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Musa, M.A.; Cooperwood, J.S.; Khan, M.O. A review of coumarin derivatives in pharmacotherapy of breast cancer. Curr. Med. Chem. 2008, 15, 2664–2679. [Google Scholar] [CrossRef] [Green Version]
- Borges, F.; Roleira, F.; Milhazes, N.; Santana, L.; Uriarte, E. Simple coumarins and analogues in medicinal chemistry: Occurrence, synthesis and biological activity. Curr. Med. Chem. 2005, 12, 887–916. [Google Scholar] [CrossRef] [PubMed]
- Geen, G.R.; Evans, J.M.; Vong, A.K.; Katritzky, A.R.; Rees, C.W.; Scriven, E.F.V. Pyrans and their Benzo Derivatives: Applications. In Comprehensive Heterocyclic Chemistry II; Lan, R., Katritzky, C.W.R., Scriven, E.F.V., Eds.; Pergamon Press: Oxford, UK, 1997; pp. 469–500. [Google Scholar]
- Egan, D.; O’Kennedy, R.; Moran, E.; Cox, D.; Prosser, E.; Thornes, R.D. The pharmacology, metabolism, analysis, and applications of coumarin and coumarin-related compounds. Drug Metab. Rev. 1990, 22, 503–529. [Google Scholar] [CrossRef] [PubMed]
- Curini, M.; Cravotto, G.; Epifano, F.; Giannone, G. Chemistry and biological activity of natural and synthetic prenyloxycoumarins. Curr. Med. Chem. 2006, 13, 199–222. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Singh, B.; Singh, N. A review on coumarins as acetylcholinesterase inhibitors for Alzheimer’s disease. Biorg. Med. Chem. 2012, 20, 1175–1180. [Google Scholar] [CrossRef]
- Liu, X.; Cole, J.M.; Waddell, P.G.; Lin, T.-C.; Radia, J.; Zeidler, A. Molecular Origins of Optoelectronic Properties in Coumarin Dyes: Toward Designer Solar Cell and Laser Applications. J. Phys. Chem. A 2011, 116, 727–737. [Google Scholar] [CrossRef]
- Barot, K.P.; Jain, S.V.; Kremer, L.; Singh, S.; Ghate, M.D. Recent advances and therapeutic journey of coumarins: Current status and perspectives. Med. Chem. Res. 2015, 24, 2771–2798. [Google Scholar] [CrossRef]
- Delogu, G.L.; Matos, M.J. Coumarins as Promising Scaffold for the Treatment of Age-related Diseases—An Overview of the Last Five Years. Curr. Top. Med. Chem. 2017, 17, 3173–3189. [Google Scholar] [CrossRef]
- Fais, A.; Corda, M.; Era, B.; Fadda, M.B.; Matos, M.J.; Quezada q, E.; Santana, L.; Picciau, C.; Podda, G.; Delogu, G. Tyrosinase Inhibitor Activity of Coumarin-Resveratrol Hybrids. Molecules 2009, 14, 2514–2520. [Google Scholar] [CrossRef] [Green Version]
- Sandhu, S.; Bansal, Y.; Silakari, O.; Bansal, G. Coumarin hybrids as novel therapeutic agents. Biorg. Med. Chem. 2014, 22, 3806–3814. [Google Scholar] [CrossRef]
- Orallo, F. Trans-resveratrol: A magical elixir of eternal youth? Curr. Med. Chem. 2008, 15, 1887–1898. [Google Scholar] [CrossRef] [PubMed]
- Fais, A.; Era, B.; Asthana, S.; Sogos, V.; Medda, R.; Santana, L.; Uriarte, E.; Matos, M.J.; Delogu, F.; Kumar, A. Coumarin derivatives as promising xanthine oxidase inhibitors. Int. J. Biol. Macromol. 2018, 120 Pt A, 1286–1293. [Google Scholar] [CrossRef]
- Stefanachi, A.; Leonetti, F.; Pisani, L.; Catto, M.; Carotti, A. Coumarin: A Natural, Privileged and Versatile Scaffold for Bioactive Compounds. Molecules 2018, 23, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, K.D.; Song, H.M.; Lee, M.J.; Pastore, M.; Anselmi, C.; De Angelis, F.; Nazeeruddin, M.K.; Gräetzel, M.; Kim, H.K. Coumarin dyes containing low-band-gap chromophores for dye-sensitised solar cells. Dyes Pigments 2011, 90, 304–310. [Google Scholar] [CrossRef]
- Nizamov, S.; Willig, K.I.; Sednev, M.V.; Belov, V.N.; Hell, S.W. Phosphorylated 3-heteroarylcoumarins and their use in fluorescence microscopy and nanoscopy. Chem. A Eur. J. 2012, 18, 16339–16348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sashidhara, K.V.; Kumar, A.; Chatterjee, M.; Rao, K.B.; Singh, S.; Verma, A.K.; Palit, G. Discovery and synthesis of novel 3-phenylcoumarin derivatives as antidepressant agents. Bioorg. Med. Chem. Lett. 2011, 21, 1937–1941. [Google Scholar] [CrossRef]
- Matos, M.J.; Terán, C.; Pérez-Castillo, Y.; Uriarte, E.; Santana, L.; Viña, D. Synthesis and Study of a Series of 3-Arylcoumarins as Potent and Selective Monoamine Oxidase B Inhibitors. J. Med. Chem. 2011, 54, 7127–7137. [Google Scholar] [CrossRef]
- Pisano, M.B.; Kumar, A.; Medda, R.; Gatto, G.; Pal, R.; Fais, A.; Era, B.; Cosentino, S.; Uriarte, E.; Santana, L.; et al. Antibacterial Activity and Molecular Docking Studies of a Selected Series of Hydroxy-3-arylcoumarins. Molecules 2019, 24, 2815. [Google Scholar] [CrossRef] [Green Version]
- Matos, M.J.; Vilar, S.; Kachler, S.; Fonseca, A.; Santana, L.; Uriarte, E.; Borges, F.; Tatonetti, N.P.; Klotz, K.N. Insight into the interactions between novel coumarin derivatives and human A3 adenosine receptors. ChemMedChem 2014, 9, 2245–2253. [Google Scholar] [CrossRef]
- Mah, S.; Jang, J.; Song, D.; Shin, Y.; Latif, M.; Jung, Y.; Hong, S. Discovery of fluorescent 3-heteroarylcoumarin derivatives as novel inhibitors of anaplastic lymphoma kinase. Org. Biomol. Chem. 2019, 17, 186–194. [Google Scholar] [CrossRef]
- Pintus, F.; Matos, M.J.; Vilar, S.; Hripcsak, G.; Varela, C.; Uriarte, E.; Santana, L.; Borges, F.; Medda, R.; Di Petrillo, A.; et al. New insights into highly potent tyrosinase inhibitors based on 3-heteroarylcoumarins: Anti-melanogenesis and antioxidant activities, and computational molecular modeling studies. Bioorg. Med. Chem. 2017, 25, 1687–1695. [Google Scholar] [CrossRef] [PubMed]
- Costas-Lago, M.C.; Besada, P.; Rodríguez-Enríquez, F.; Viña, D.; Vilar, S.; Uriarte, E.; Borges, F.; Terán, C. Synthesis and structure-activity relationship study of novel 3-heteroarylcoumarins based on pyridazine scaffold as selective MAO-B inhibitors. Eur. J. Med. Chem. 2017, 139, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.J.; Varela, C.; Vilar, S.; Hripcsak, G.; Borges, F.; Santana, L.; Uriarte, E.; Fais, A.; Di Petrillo, A.; Pintus, F.; et al. Design and discovery of tyrosinase inhibitors based on a coumarin scaffold. RSC Adv. 2015, 5, 94227–94235. [Google Scholar] [CrossRef]
- Lin, H.-C.; Tsai, S.-H.; Chen, C.-S.; Chang, Y.-C.; Lee, C.-M.; Lai, Z.-Y.; Lin, C.-M. Structure–activity relationship of coumarin derivatives on xanthine oxidase-inhibiting and free radical-scavenging activities. Biochem. Pharmacol. 2008, 75, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Pramod, A.G.; Renuka, C.G.; Nadaf, Y.F. Electronic Structure, Optical Properties and Quantum Chemical Investigation on Synthesized Coumarin Derivative in Liquid Media for Optoelectronic Devices. J. Fluoresc. 2019, 29, 953–968. [Google Scholar] [CrossRef] [PubMed]
- Kohn, W. Nobel Lecture: Electronic structure of matter—Wave functions and density functionals. Rev. Mod. Phys. 1999, 71, 1253–1266. [Google Scholar] [CrossRef] [Green Version]
- Azad, I.; Akhter, Y.; Khan, T.; Azad, M.I.; Chandra, S.; Singh, P.; Kumar, D.; Nasibullah, M. Synthesis, quantum chemical study, AIM simulation, in silico ADMET profile analysis, molecular docking and antioxidant activity assessment of aminofuran derivatives. J. Mol. Struct. 2019, 127285. [Google Scholar] [CrossRef]
- Molteni, E.; Cappellini, G.; Onida, G.; Mula, G. Extensive stacking of DHI-like monomers as a model of out-of-plane complexity in eumelanin protomolecules: Chemical and structural sensitivity of optical absorption spectra. Chem. Phys. 2019, 524, 92–100. [Google Scholar] [CrossRef]
- Zhao, D.; Lu, Q.; Su, R.; Li, Y.; Zhao, M. Light harvesting and optical-electronic properties of two quercitin and rutin natural dyes. Appl. Sci. 2019, 9, 2567. [Google Scholar] [CrossRef] [Green Version]
- Marques, M.A.L.; Gross, E.K.U. Time-Dependent Density Functional Theory. Annu. Rev. Phys. Chem. 2004, 55, 427–455. [Google Scholar] [CrossRef] [Green Version]
- Basavarajappa, K.V.; Arthoba Nayaka, Y.; Purushothama, H.T.; Vinaya, M.M.; Antony, A.; Poornesh, P. Optoelectronic and current-voltage studies for novel coumarin dyes. Int. J. Environ. Anal. Chem. 2019, 1–14. [Google Scholar] [CrossRef]
- Hara, K.; Sato, T.; Katoh, R.; Furube, A.; Ohga, Y.; Shinpo, A.; Suga, S.; Sayama, K.; Sugihara, H.; Arakawa, H. Molecular design of coumarin dyes for efficient dye-sensitized solar cells. J. Phys. Chem. B 2003, 107, 597–606. [Google Scholar] [CrossRef]
- Oprea, C.I.; Panait, P.; Cimpoesu, F.; Ferbinteanu, M.; Girtu, M.A. Density functional theory (DFT) study of coumarin-based dyes adsorbed on TiO(2) nanoclusters-applications to dye-sensitized solar cells. Materials 2013, 6, 2372–2392. [Google Scholar] [CrossRef] [Green Version]
- Hara, K.; Wang, Z.-S.; Sato, T.; Furube, A.; Katoh, R.; Sugihara, H.; Dan-oh, Y.; Kasada, C.; Shinpo, A.; Suga, S. Oligothiophene-containing coumarin dyes for efficient dye-sensitized solar cells. J. Phys. Chem. B 2005, 109, 15476–15482. [Google Scholar] [CrossRef]
- Sánchez-de-Armas, R.; San Miguel, M.Á.; Oviedo, J.; Sanz, J.F. Coumarin derivatives for dye sensitized solar cells: A TD-DFT study. Phys. Chem. Chem. Phys. 2012, 14, 225–233. [Google Scholar] [CrossRef]
- Mutlak, F.; Mohi, A.; Alwan, T. Density functional theory study of molecular structure, Electronic properties, UV–Vis spectra on coumarin102. Baghdad Sci. J. 2016, 13, 143–152. [Google Scholar] [CrossRef]
- Kumar, A.; Cappellini, G.; Delogu, F. Electronic and optical properties of chromophores from hexeneuronic acids. Cellulose 2019, 26, 1489–1501. [Google Scholar] [CrossRef]
- Liu, X.; Cole, J.M.; Low, K.S. Solvent Effects on the UV–vis Absorption and Emission of Optoelectronic Coumarins: A Comparison of Three Empirical Solvatochromic Models. J. Phys. Chem. C 2013, 117, 14731–14741. [Google Scholar] [CrossRef]
- Pramod, A.G.; Renuka, C.G.; Nadaf, Y.F.; Rajaramakrishna, R. Impact of solvents on energy gap, photophysical, photometric properties for a new class of 4-HCM coumarin derivative: Nonlinear optical studies and optoelectronic applications. J. Mol. Liq. 2019, 292, 111383. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Rassolov, V.A.; Ratner, M.A.; Pople, J.A.; Redfern, P.C.; Curtiss, L.A. 6-31G* basis set for third-row atoms. J. Comput. Chem. 2001, 22, 976–984. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Chermette, H. Chemical reactivity indexes in density functional theory. J. Comput. Chem. 1999, 20, 129–154. [Google Scholar] [CrossRef]
- Kumar, A.; Gatto, G.; Delogu, F.; Pilia, L. DFT study of [Pt(Cl)2L] complex (L = rubeanic acid) and its derived compounds with DNA purine bases. Chem. Phys. 2020, 530, 110646. [Google Scholar] [CrossRef]
- Kumar, A.; Cardia, R.; Cappellini, G. Electronic and optical properties of chromophores from bacterial cellulose. Cellulose 2018, 25, 2191–2203. [Google Scholar] [CrossRef]
- Cardia, R.; Malloci, G.; Mattoni, A.; Cappellini, G. Effects of TIPS-Functionalization and Perhalogenation on the Electronic, Optical, and Transport Properties of Angular and Compact Dibenzochrysene. J. Phys. Chem. A 2014, 118, 5170–5177. [Google Scholar] [CrossRef]
- Jones, R.O.; Gunnarsson, O. The density functional formalism, its applications and prospects. Rev. Mod. Phys. 1989, 61, 689–746. [Google Scholar] [CrossRef]
- Malloci, G.; Cappellini, G.; Mulas, G.; Satta, G. Quasiparticle effects and optical absorption in small fullerenelike GaP clusters. Phys. Rev. B 2004, 70, 205429. [Google Scholar] [CrossRef]
- Cardia, R.; Malloci, G.; Rignanese, G.M.; Blase, X.; Molteni, E.; Cappellini, G. Electronic and optical properties of hexathiapentacene in the gas and crystal phases. Phys. Rev. B 2016, 93, 235132. [Google Scholar] [CrossRef]
- Molteni, E.; Cappellini, G.; Onida, G.; Fratesi, G. Optical properties of organically functionalized silicon surfaces: Uracil-like nucleobases on Si(001). Phys. Rev. B 2017, 95, 075437. [Google Scholar] [CrossRef] [Green Version]
- Valiev, M.; Bylaska, E.J.; Govind, N.; Kowalski, K.; Straatsma, T.P.; Van Dam, H.J.J.; Wang, D.; Nieplocha, J.; Apra, E.; Windus, T.L.; et al. NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 2010, 181, 1477–1489. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Thompson, M.A. ArgusLab 4.0.1; Planaria Software LLC: Seattle, WA, USA, 2004. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Chakravarty, H.; Bal, N.C.; Balaraju, T.; Jena, N.; Misra, G.; Bal, C.; Pieroni, E.; Periasamy, M.; Sharon, A. Identification of calcium binding sites on calsequestrin 1 and their implications for polymerization. Mol. Biosyst. 2013, 9, 1949–1957. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Sechi, L.A.; Caboni, P.; Marrosu, M.G.; Atzori, L.; Pieroni, E. Dynamical insights into the differential characteristics of Mycobacterium avium subsp. paratuberculosis peptide binding to HLA-DRB1 proteins associated with multiple sclerosis. New J. Chem. 2015, 39, 1355–1366. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Linstrom, P.J.; Mallard, W.G. NIST Chemistry WebBook; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2018; p. 20899.
- Johnson, R.D., III. NIST Computational Chemistry Comparison and Benchmark Database, Release 20; U.S. Secretary of Commerce: Washington, DC, USA, 2019.
- Alam, M.; Alam, M.J.; Azaz, S.; Parveen, M.; Park, S.; Ahmad, S. DFT/TD-DFT calculations, spectroscopic characterizations (FTIR, NMR, UV–vis), molecular docking and enzyme inhibition study of 7-benzoyloxycoumarin. Comput. Biol. Chem. 2018, 73, 65–78. [Google Scholar] [CrossRef]
- Munshi, P.; Guru Row, T.N. Exploring the Lower Limit in Hydrogen Bonds: Analysis of Weak C−H···O and C−H···π Interactions in Substituted Coumarins from Charge Density Analysis. J. Phys. Chem. A 2005, 109, 659–672. [Google Scholar] [CrossRef]
- Redchenko, V.V.; Safronov, A.I.; Kirpichenok, M.A.; Grandberg, I.I.; Traven’, V.F. Electronic structure of π-systems. XV. Photoelectron spectra of 7-aminocoumarin derivatives. J. Gen. Chem. USSR 1992, 62, 2313. [Google Scholar]
- Bauzá, A.; Quiñonero, D.; Deyà, P.M.; Frontera, A. Is the use of diffuse functions essential for the properly description of noncovalent interactions involving anions? J. Phys. Chem. A 2013, 117, 2651–2655. [Google Scholar] [CrossRef] [PubMed]
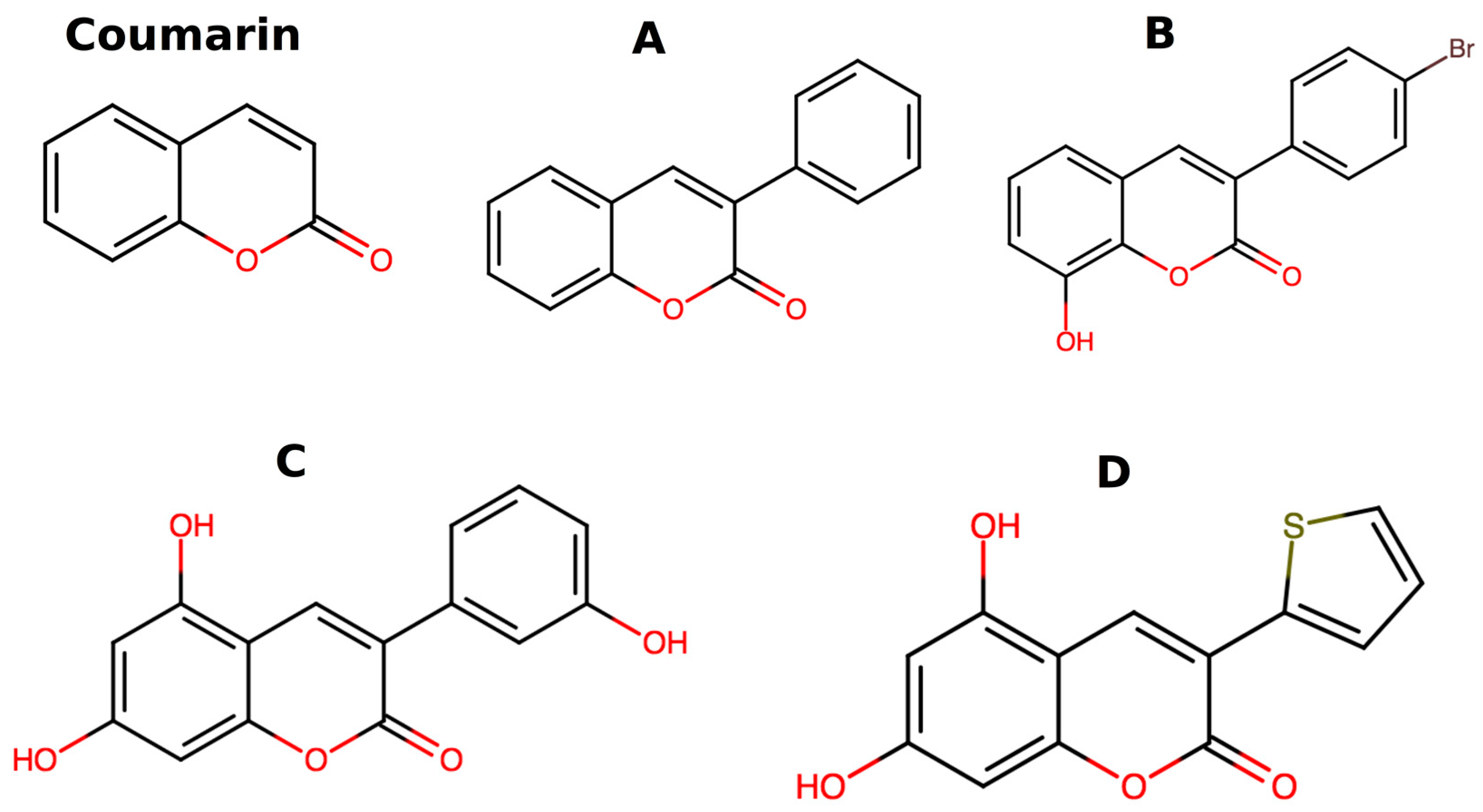
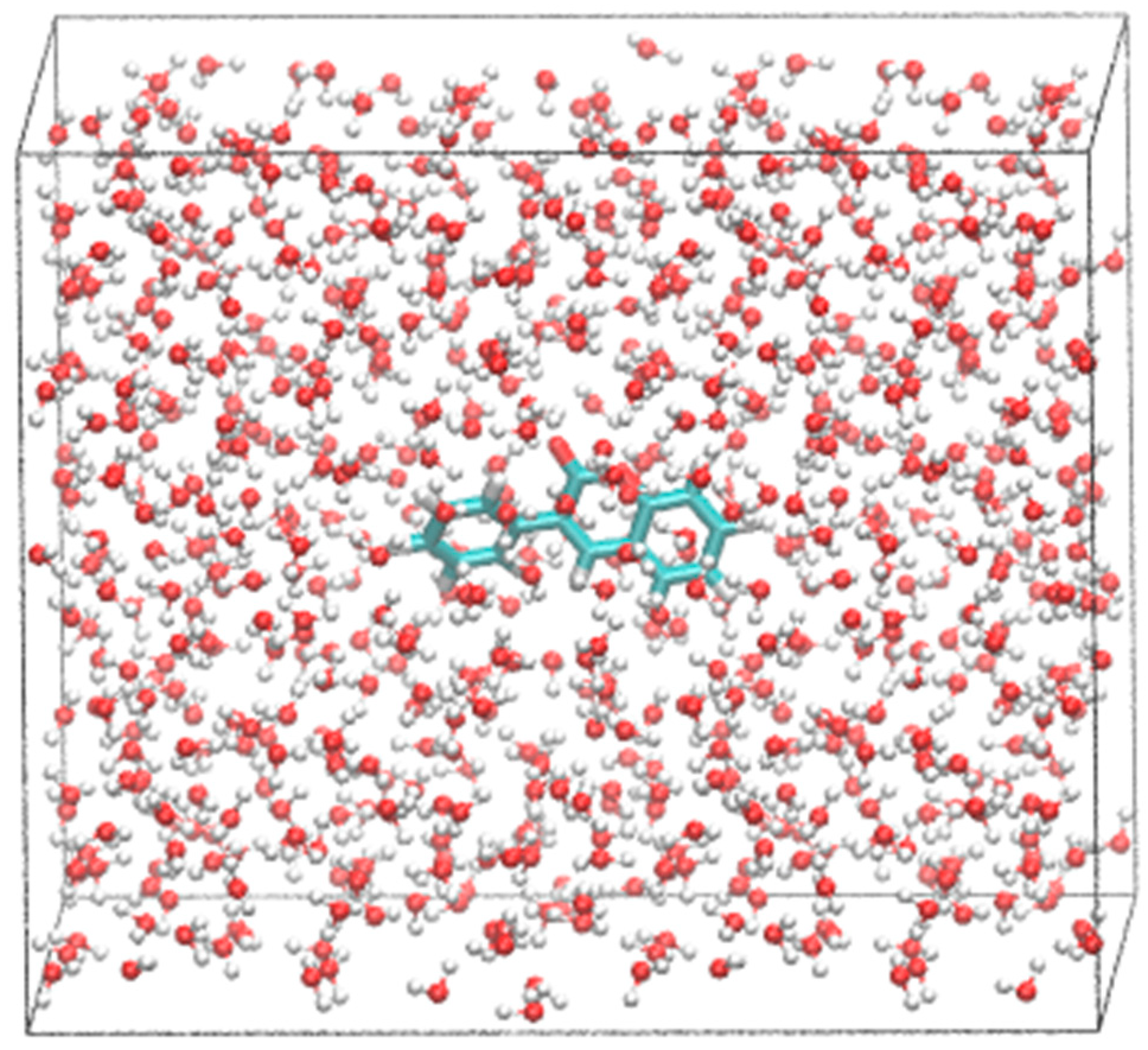
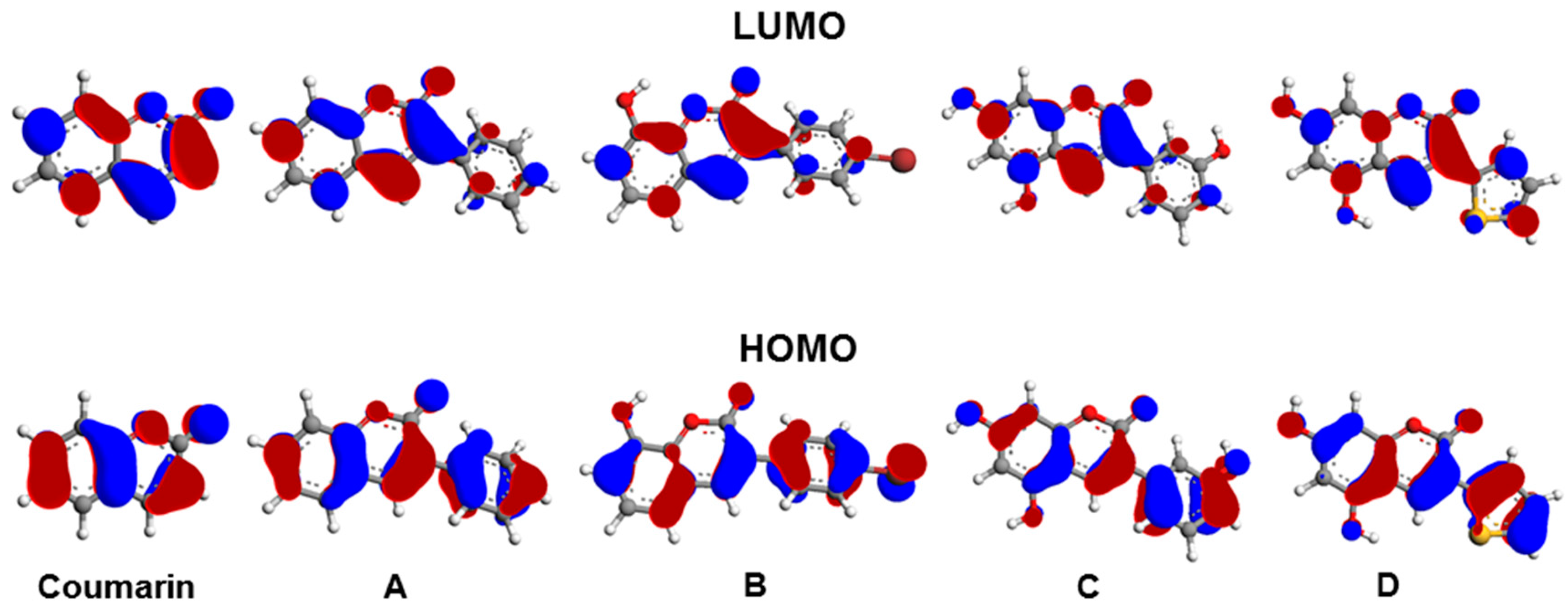


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
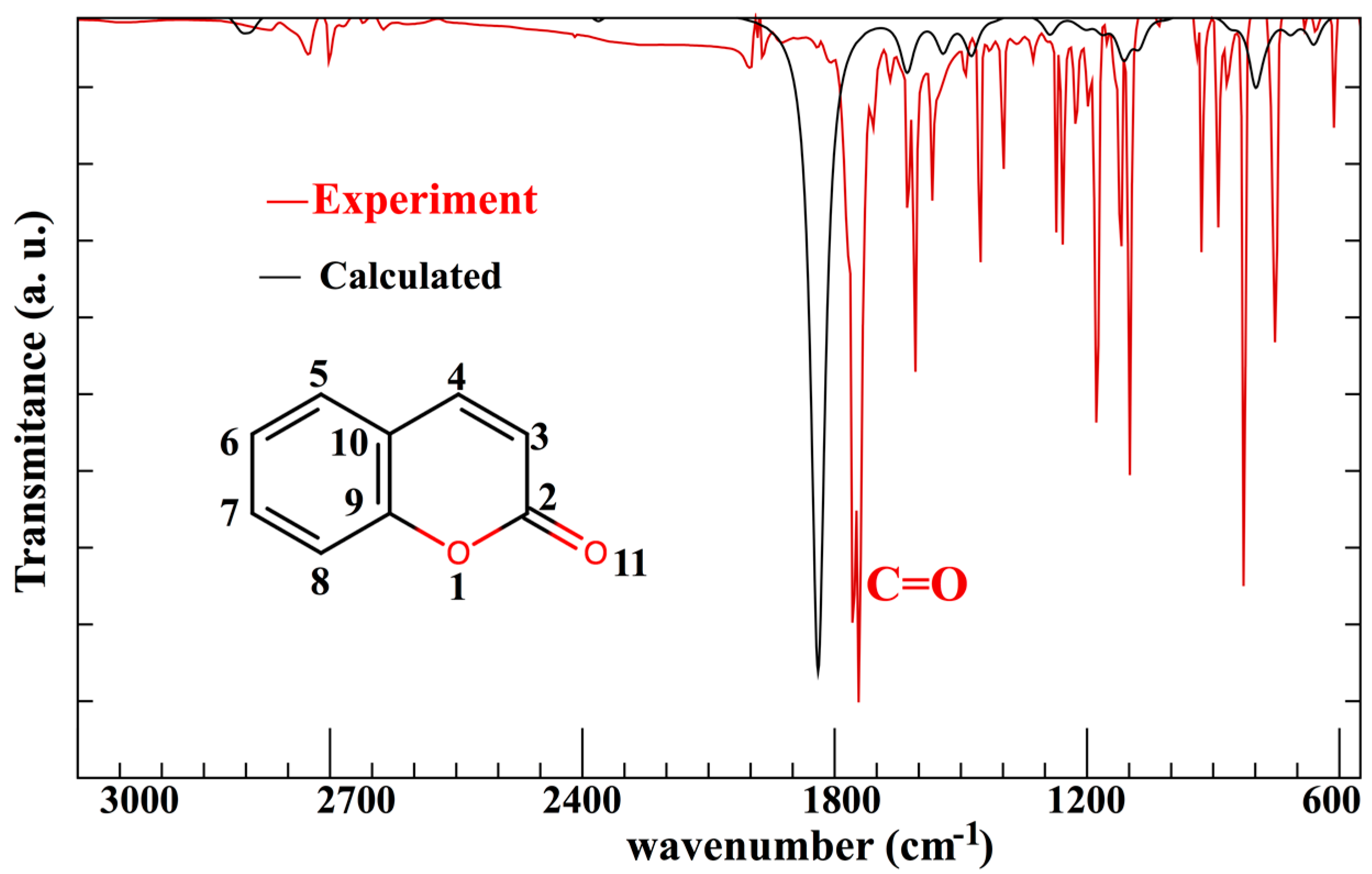
| Atom 1 | Atom 2 | Exp.≠ | B3LYP/6-31G* | B3LYP/6-311+G* | PBE/6-31G* |
|---|---|---|---|---|---|
| 1 O | 2 C | 1.371 | 1.398 (1.97%) | 1.396 (1.82%) | 1.419 (3.5%) |
| 1 O | 9 C | 1.372 | 1.365 (−0.5%) | 1.365 (−0.5%) | 1.367 (−0.36%) |
| 2 C | 11 O | 1.216 | 1.208 (−0.65%) | 1.202 (−1.15%) | 1.217 (0.08%) |
| 2 C | 3 C | 1.454 | 1.460 (0.4%) | 1.459 (0.34%) | 1.459 (0.34%) |
| 3 C | 4 C | 1.352 | 1.352 (0%) | 1.349 (−0.22%) | 1.363 (0.81%) |
| 4 C | 10 C | 1.440 | 1.442 (0.13%) | 1.440 (0%) | 1.441 (0.07%) |
| 5 C | 10 C | 1.406 | 1.407 (0.07%) | 1.406 (0%) | 1.414 (0.57%) |
| 5 C | 6 C | 1.388 | 1.388 (0%) | 1.385 (−0.22%) | 1.394 (0.432%) |
| 6 C | 7 C | 1.402 | 1.403 (0.07%) | 1.402 (0%) | 1.402 (0%) |
| 7 C | 8 C | 1.390 | 1.391 (−0.07%) | 1.389 (−0.07%) | 1.397 (0.50%) |
| 8 C | 9 C | 1.394 | 1.396 (0.14%) | 1.394 (0%) | 1.404 (0.72%) |
| 9 C | 10 C | 1.400 | 1.408 (0.57%) | 1.405 (0.36%) | 1.419 (1.36%) |
| Compound | Coumarin | A | B | C | D | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| EHOMO (eV) | −6.50 | −6.87 | −6.07 | −6.45 | −6.26 | −6.59 | −5.61 | −6.04 | −5.58 | −5.97 |
| ELUMO (eV) | −1.88 | −2.28 | −2.04 | −2.32 | −2.23 | −2.58 | −1.71 | −2.14 | −2.04 | −2.48 |
| η = (ELUMO − EHOMO)/2 (eV) | 2.31 | 2.30 | 2.01 | 2.07 | 2.01 | 2.01 | 1.95 | 1.95 | 1.77 | 1.75 |
| CP = (ELUMO + EHOMO)/2 (eV) | −4.19 | −4.58 | −4.05 | −4.39 | −4.25 | −4.59 | −3.66 | −4.09 | −3.81 | −4.23 |
| Ionization energy IEV (eV) | 8.41 | 8.71 ≠ | 7.64 | 7.96 | 7.72 | 7.98 | 7.11 | 7.48 | 7.14 | 7.46 |
| Electron affinity EAV (eV) | −0.04 | 0.49 | 0.35 | 0.80 | 0.67 | 1.09 | 0.15 | 0.65 | 0.48 | 1.00 |
| Quasi particle gap EQP (eV) | 8.45 | 8.22 | 7.29 | 7.16 | 7.05 | 6.89 | 6.96 | 6.83 | 6.66 | 6.46 |
| Compound | Coumarin | A | B | C | D | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Properties | PBE | Δ | PBE | Δ | PBE | Δ | PBE | Δ | PBE | Δ |
| EHOMO (eV) | −5.72 | −0.78 | −5.39 | −0.68 | −5.51 | −0.75 | −5.01 | −0.6 | −4.88 | −0.7 |
| ELUMO (eV) | −2.60 | 0.72 | −2.66 | 0.62 | −2.89 | 0.66 | −2.53 | 0.82 | −2.63 | 0.59 |
| (ELUMO − EHOMO)/2 (eV) | 1.56 | 0.75 | 1.37 | 0.64 | 1.31 | 0.7 | 1.24 | 0.71 | 1.13 | 0.64 |
| CP = (ELUMO + EHOMO)/2 (eV) | −4.16 | −0.03 | −4.03 | −0.02 | −4.2 | −0.05 | −3.77 | 0.11 | −3.76 | −0.05 |
| Ionization energy IEV (eV) | 8.28 | 0.13 | 7.47 | 0.17 | 7.42 | 0.3 | 6.98 | 0.13 | 6.93 | 0.21 |
| Electron affinity EAV (eV) | 0.07 | −0.11 | 0.51 | −0.16 | 0.80 | −0.13 | 0.41 | −0.26 | 0.54 | −0.06 |
| Quasi particle gap EQP (eV) | 8.21 | 0.24 | 6.96 | 0.33 | 6.62 | 0.43 | 6.57 | 0.39 | 6.39 | 0.27 |
| Molecule | B3LYP/6-31G* | B3LYP/6-311+G* | PBE/6-31+G* | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Main Peak | EOPT | Main Peak | EOPT | Main Peak | EOPT | |||||||
| eV | OS | eV | OS | eV | OS | eV | OS | eV | OS | eV | OS | |
| A | 3.80 | 0.50 | 3.80 | 0.50 | 3.77 | 0.49 | 3.77 | 0.49 | 6.47 | 0.38 | 3.37 | 0.35 |
| B | 3.72 | 0.55 | 3.59 | 0.02 | 3.66 | 0.48 | 3.58 | 0.06 | 3.16 | 0.34 | 2.92 | 0.02 |
| C | 3.82 | 0.36 | 3.50 | 0.21 | 5.89 | 0.38 | 3.47 | 0.23 | 6.32 | 0.14 | 2.74 | 0.04 |
| D | 3.35 | 0.58 | 3.35 | 0.58 | 3.24 | 0.55 | 3.24 | 0.55 | 2.98 | 0.42 | 2.98 | 0.42 |
| Molecule | B3LYP/6-31G* | B3LYP/6-311+G* | PBE/6-31+G* | |||
|---|---|---|---|---|---|---|
| (eV) | EOPT | EBIND | EOPT | EBIND | EOPT | EBIND |
| A | 3.80 | 3.49 | 3.77↓ | 3.36 ↓ (3.4%) | 3.37 ↓ | 3.59 ↑ (2.9%) |
| B | 3.59 | 3.46 | 3.58 ↓ | 3.31 ↓ (4.3%) | 2.92 ↓ | 3.70 ↑ (6.9%) |
| C | 3.50 | 3.46 | 3.47 ↓ | 3.33 ↓ (3.8%) | 2.74 ↓ | 3.83 ↑ (10.7%) |
| D | 3.35 | 3.31 | 3.24 ↓ | 3.11 ↓ (6%) | 2.98 ↓ | 3.41 ↑ (3%) |
| Molecule | B3LYP/6-31G* (Vacuum) | B3LYP/6-31G* (Water) | ||||
|---|---|---|---|---|---|---|
| MP (eV) | OS | Transition | MP (eV) | OS | Transition | |
| A | 3.80 | 0.5 | H→L (96.3%) | 3.46 | 0.54 | H→L (98%) |
| B | 3.72 | 0.55 | H→L (82.6%) | 3.38 | 0.65 | H→L (98%) |
| C | 3.82 | 0.36 | H–1→L (79.4%) | 3.64 | 0.49 | H-1→L (81%) |
| D | 3.35 | 0.58 | H →L (97. 8%) | 3.31 | 0.44 | H→L (97%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, A.; Baccoli, R.; Fais, A.; Cincotti, A.; Pilia, L.; Gatto, G. Substitution Effects on the Optoelectronic Properties of Coumarin Derivatives. Appl. Sci. 2020, 10, 144. https://doi.org/10.3390/app10010144
Kumar A, Baccoli R, Fais A, Cincotti A, Pilia L, Gatto G. Substitution Effects on the Optoelectronic Properties of Coumarin Derivatives. Applied Sciences. 2020; 10(1):144. https://doi.org/10.3390/app10010144
Chicago/Turabian StyleKumar, Amit, Roberto Baccoli, Antonella Fais, Alberto Cincotti, Luca Pilia, and Gianluca Gatto. 2020. "Substitution Effects on the Optoelectronic Properties of Coumarin Derivatives" Applied Sciences 10, no. 1: 144. https://doi.org/10.3390/app10010144
APA StyleKumar, A., Baccoli, R., Fais, A., Cincotti, A., Pilia, L., & Gatto, G. (2020). Substitution Effects on the Optoelectronic Properties of Coumarin Derivatives. Applied Sciences, 10(1), 144. https://doi.org/10.3390/app10010144