Featured Application
Pt/TiO2 catalysts are widely applied in catalysis, especially in oxidation reactions within exhaust gas after treatment. Novel synthetic approaches may significantly improve their catalytic properties. Pulsed laser ablation in liquid offers altered properties of oxide nanopowders such as enhancement of thermostability and increased concentration of defects providing strong interaction with Pt.
Abstract
A set of physicochemical methods, including X-ray photoelectron spectroscopy (XPS), X-ray diffraction, electron microscopy and X-ray absorption spectroscopy, was applied to study Pt/TiO2 catalysts prepared by impregnation using a commercial TiO2-P25 support and a support produced by pulsed laser ablation in liquid (PLA). The Pt/TiO2-PLA catalysts showed increased thermal stability due to the localization of the highly dispersed platinum species at the intercrystalline boundaries of the support particles. In contrast, the Pt/TiO2-P25 catalysts were characterized by uniform distribution of the Pt species over the support. Analysis of Pt4f XP spectra shows that oxidized Pt2+ and Pt4+ species are formed in the Pt/TiO2-P25 catalysts, while the platinum oxidation state in the Pt/TiO2-PLA catalysts is lower due to stronger interaction of the active component with the support due to strong interaction via Pt-O-Ti bonds. The Pt4f XP spectra of the samples after reaction show Pt2+ and metallic platinum, which is the catalytically active species. The study of the catalytic properties in ammonia oxidation showed that, unlike the catalysts prepared with a commercial support, the Pt/TiO2-PLA samples show higher stability during catalysis and significantly higher selectivity to N2 in a wide temperature range of 200–400 °C.
1. Introduction
Platinum is one of the most widely used active components of catalysts for a wide range of oxidative reactions. Platinum catalysts are used for complete oxidation of CO, soot and hydrocarbons [1,2], epoxidation [3], in fuel cells [4] and selective oxidation [5,6]. Most commonly, platinum is used in supported form. Depending on the type of the catalytic process, different supports are chosen: carbon materials [7], inert oxides such as SiO2 [8] or Al2O3 [9], reducible oxides, e.g., CeO2 [10], Fe2O3 [11], TiO2 [12]. When using reducible oxide supports a high degree of the platinum interaction with the support (strong metal-support interaction, SMSI) can be achieved. This may result in formation of Pt particles of specific morphology, clusters, ionic Pt forms stabilized in the support oxide lattice [13], or isolated platinum sites on the surface [14].
For many reactions an increased dispersion of the active component helps improving activity of the catalysts [15]. However, in some cases, high dispersion of the active component is not beneficial [16]. For instance, selective oxidation of ammonia to N2 (ammonia slip removal) proceeds most efficiently on platinum nanoparticles with sizes of at least 2 and up to 10 nm [9,17] supported on aluminum, niobium, silicon or titanium oxides. The latter is known for its strong interaction with platinum when heated in reducing atmosphere. The Pt/TiO2 interaction, on the one hand, can stabilize Pt particles on the surface. On the other hand, it might result in Pt encapsulation by the support, preventing adsorption of reagents [18,19]. It is also known that at high temperatures a phase transformation of TiO2 from anatase to rutile can occur leading to a loss of the catalyst activity [20].
For industrial applications, catalyst stability is required, so thermal stability of the active component is of high importance. Using TiO2 as a support might help to stabilize platinum particles of required sizes and oxidation states on the surface due to the SMSI. The metal-support interaction is ensured by defects of the support surface. Therefore, the most suitable synthesis methods are those which use nonequilibrium processes to produce structures with high density of defects: burning a precursor solution (solution-combustion) [21,22], spraying in a flame (spray pyrolysis) [23,24], atomization in electric discharge [25,26] or laser evaporation [27,28,29]. Pulsed laser ablation (PLA) in liquid phase is a simple and environmentally friendly method for obtaining dispersions of particles with a high density of defects. The pulsed laser ablation (PLA)-prepared dispersions of nanoparticles can be further used for the synthesis of powder catalysts. The efficiency of the PLA technique for preparation of powder catalysts was previously shown for a number of catalytic systems based on ceria, alumina, and iron oxides in which either the support nanoparticles (NPs) or Pt NPs were prepared by the PLA [30,31,32].
The aim of this work was to study the influence of the TiO2 support synthesized by the PLA technique on the catalytic performance of Pt/TiO2 catalysts during selective oxidation of ammonia to N2 (ammonia slip catalysts, ASCs).
2. Materials and Methods
2.1. Supports Preparation
The first type of support was prepared by pulsed laser ablation (PLA). The fundamental harmonic radiation of a pulsed nanosecond Nd: YAG laser LS-2132M, LOTIS TII (wavelength of 1064 nm, pulse energy up to 200 mJ, 7 ns, 20 Hz) was used. A titanium metal plate (99.99 at.% purity) mounted on a movable mount, which automatically scans the laser beam over the surface, was used as a target. The movement of the target material relative to the laser beam prevents formation of caverns and craters. This ensures stable focusing of the laser beam and eliminates separation of macroscopic particles from the target. Distilled water was used as a solvent. Ablation was carried out in a cylindrical glass reactor. The initial volume of the solvent was 100 mL. The laser beam was focused by an f = 50 mm quartz lens and brought through the side wall of the vessel, which ensured constant focusing and absence of spatter. The pulsed radiation power density was 0.25–0.5 GW/cm2. The resulting dispersion was kept for ~ 10 h. The resulting dispersion was dried in air at 60 °C; and the obtained powder was calcined at 400 °C for 4 h in air. Titanium oxide prepared by PLA was denoted TiOx-PLA.
For comparison, a commercial titanium oxide “AEROXIDE®® TiO2 P25” (Evonik), denoted TiO2-P25, was used as the second support. It was not subjected to any additional treatments before its use for the synthesis of catalysts.
2.2. Catalysts Preparation
Pt/TiO2 catalysts with a platinum content of 2 wt.% were prepared by incipient wetness impregnation. A solution of commercial platinum nitrate Pt(NO3)4, with platinum content of 17.87 wt.% (KZCM) was used as platinum precursor. The titanium oxides TiOx-PLA and TiO2-P25 were used as supports. Before synthesis, the supports were dried in air for 2 h at 300 °C. The products were dried to an air-dry state for at least 16 h, then for 1 h at 60 °C and 2 h at 120 °C. Afterwards the catalysts were placed in crucibles and heated in a muffle furnace in air at 400 °C for 4 h (heating rate 5 °C/min). The resulting catalysts were denoted Pt/Ti-PLA-400 and Pt/Ti-P25-400, respectively. Samples were calcined using similar procedures at 600 and 800 °C to produce Pt/Ti-PLA-600, Pt/Ti-PLA-800, Pt/Ti-P25-600, Pt/Ti-P25-800. Concentration of platinum in the solution and in the final catalysts was determined using an Optima 430 DV optical atomic emission spectrometer (Perkin Elmer, Waltham, MA, USA).
2.3. X-ray Phase Analysis (XRD)
X-ray diffraction patterns were obtained on a Bruker D8 instrument (Germany) using CuKα radiation. The signal was recorded using a one-dimensional LynxEye detector. The survey was carried out in the range of angles 2θ = 15–90°, step 0.05°, 5 sec time per point., The ICDD PDF-2 powder database was used for phase analysis. The structure refinement and profile analysis were performed in the TOPAS software package [33]. Reflections of Pt/TiO2 samples were analyzed and fitted by the Rietveld method as a set of different TiO2 phases with fixed parameters (determined in the study of pure supports) and a platinum metal phase. To describe the instrumental profile, an external standard was used—silicon metal powder, recorded under identical conditions. The size of the coherent scattering region (CSR) was calculated by the LVol-IB value.
2.4. X-ray Absorption Spectroscopy (XANES, EXAFS)
X-ray absorption near edge structure (XANES) spectra at the Pt L3 absorption edge were recorded ex situ at the P65 beamline of the PETRA III synchrotron radiation source (DESY, Hamburg, Germany) in transmission mode. The energy of the X-rays was selected using a Si(111) double crystal monochromator, higher harmonics rejection was performed using a pair of Rh-coated Si mirrors bent at 2.5 mrad, and the beam size was selected by slits as 0.4 (vert.) × 1.5 (hor.) mm2. Catalyst samples were measured as 100–200 μm sieved powders packed in quartz capillaries (o.d. = 1.5 mm, 0.02 mm wall thickness). The spectra were corrected for the energy shift using a spectrum of Pt foil measured simultaneously and then normalized using the Athena program from the IFFEFIT software package [34]. For the analysis of extended X-ray absorption fine structure (EXAFS) the spectra were background-subtracted, k2-weighted and Fourier-transformed in the k-range 2.5–11.0 Å−1 and multiplied by a Hanning window with sill size of 1 Å−1. The amplitude reduction factor S02 = 0.74 was obtained by fitting the Pt foil and the PtO2 (Alfa Aesar, 99.95%) reference spectra to structural models as reported in the Inorganic Crystal Structure Database (ICSD, CCs = 64,923 and 4415). The fits were performed on k1, k2, and k3-weighted data using Artemis [34] by a least square method in R-space between 1.0 and 3.0 Å. Coordination numbers, interatomic distances, energy shift (δE0) and mean square deviation of interatomic distances (σ2) were refined during the fitting. The absolute misfit between theory and experiment was expressed by ρ.
2.5. X-ray Photoelectron Spectroscopy (XPS)
XPS analysis of the samples was performed using an ES-300 spectrometer (KRATOS Analytical). Spectra were recorded using non-monochromatized MgKα radiation (1253.6 eV). The spectrometer was calibrated using Au4f7/2 and Cu2p3/2 lines with binding energies of 84.0 and 932.7 eV for metal foils of gold and copper, respectively. A titanium Ti2p3/2 line with a maximum at a binding energy 458.6 eV [35] was used as an internal standard. The position of the C1s peak was 285.1 eV, which corresponds to the characteristic values for amorphous carbon and hydrocarbons on the surface of the oxides. To analyze quantitative composition of the samples and the chemical state of the elements on the surface, the core-level spectra of the elements (Ti2p, Pt4f, C1s, O1s) were recorded. The spectra were fitted with the individual components using the Gauss and Lorentz functions after Shirley background subtraction. The spectra were processed using XPS-Calc software tested previously on model catalytic systems [36] and dispersed powder catalysts [13].
2.6. High Resolution Transmission Electron Microscopy (HRTEM)
The data were collected on a JEM-2200FS electron microscope (JEOL Ltd., Japan) with an accelerating voltage of 200 kV. This allowed obtaining high-resolution transmission electron microscopy (HRTEM) and high-angle annular dark-field imaging scanning transmission electron microscopy (HAADF STEM) images with a spatial resolution of 1 Å. Lattice fringes obtained by high resolution transmission electron microscopy were analyzed by the Fourier method. The samples were dispersed in ethanol ultrasonically and deposited by sputtering the ethanol dispersion on 3 mm copper grids covered with a carbon film. Particle size distribution was determined from the TEM data using “ImageJ” software [37].
2.7. Raman Spectroscopy
Raman spectra of the powder samples were obtained using an InVia (Renishaw, UK) confocal Raman dispersive spectrometer equipped with a Leica microscope with a 50×objective. Excitation was performed with a 532 nm 100 mW semiconductor laser. To prevent sample heating and phase transformation [30] only 0.5% of laser power was employed with the 100% defocusing mode. The Raman spectra were measured in the 100–1000 cm−1 range with a 1 cm−1 spectral resolution.
2.8. Catalytic Activity Measurements
The redox properties of the samples and their catalytic activity in ammonia oxidation were studied by temperature-programmed reaction (TPR-NH3+O2) and temperature-programmed reduction (TPR-H2). The experiments were carried out in an automatic setup equipped with a plug flow quartz reactor with a FTIR spectrometer (I1801, MIDAC corp., USA) and a gas chromatograph (Crystal 2000 M, CHROMATEC). The reaction mixture containing 0.1 vol.% NH3, 4.0 vol.% O2 (balance He) was introduced at a rate of 500 cm3/min and GHSV–120 000 h−1. Each sample (0.145 g) was heated twice in the NH3+O2 mixture from room temperature to 400 °C. Analysis of H2 consumption was performed using mass spectrometry (QMS200 high pressure sampling system, Stanford Research Systems, Sunnyvale, CA, USA). The setup description and experimental conditions are presented in more detail in [6].
3. Results
3.1. XRD Analysis
Phase composition of the samples was determined by XRD. Figure 1 shows X-ray diffraction patterns of the samples depending on the calcination temperature. All XRD patterns were refined by the Rietveld method. Lattice parameters for all observed phases remain practically unchanged with the variation of the calcination temperature and, within the error bars, correspond to anatase (ICDD PDF-2 # 21-1272), rutile (ICDD PDF-2 # 21-1276) and brookite (ICDD PDF -2 # 29-1360) phases (Table A1).
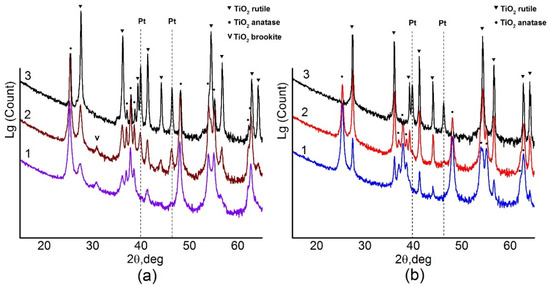
Figure 1.
(a) X-ray diffraction (XRD) data for Pt/Ti-PLA-400 (1), Pt/Ti-PLA -600 (2) and Pt/Ti-PLA -800 (3); (b) XRD data for catalysts Pt/Ti-P25-400 (1), Pt/Ti-P25-600 (2) and Pt/Ti-P25-800 (3).
X-ray diffraction data show that the Pt/Ti-PLA-400 catalyst contains a mixture of TiO2 phases including anatase (66%), rutile (19%) and brookite (15%) (Table 1). The Pt/Ti-P25-400 catalyst contains only two titanium dioxide phases: anatase (84%) and rutile (16%) (Table 1). An increase of the calcination temperature leads to an increase of the amount of the more stable TiO2-rutile phase. Only the TiO2-rutile phase is observed in the Pt/Ti-P25-800 sample. For the Pt/Ti-PLA samples, calcination at 600 °C leads to disappearance of the TiO2-brookite phase but the TiO2-anatase phase remains stable even after calcination at 800 °C.

Table 1.
Phase composition and percentage in Pt/Ti-PLA and Pt/Ti-P25 samples by XRD data.
Table 1 shows that, for the catalysts calcined at 400 °C, crystallite sizes of the TiO2-anatase phase, estimated from the coherent scattering region (D), are quite similar: 19–22 nm. The surface areas of the Pt/Ti-PLA-400 and Pt/Ti-P25-400 samples are also similar: 79 and 64 m2/g, respectively. However, in the case of PLA-produced support the rutile particles are significantly smaller: 7 nm compared to 29 nm. The brookite phase has an even lower D value of about 5 nm. An increase in the calcination temperature leads to an increase in the D values for the TiO2-rutile and TiO2-anatases phases for all samples, but the commercial support is more prone to sintering. Thus, the support obtained by the PLA method is more thermally stable with respect to phase transitions and sintering.
No reflections corresponding to Pt phases were observed for the samples calcined at 400 °C implying that the samples contain platinum in a highly dispersed or an amorphous state. Metallic Pt phase (ICDD PDF-2 # 04-0802) is observed upon calcination of the samples at 600 °C and higher. The crystallite sizes of metallic Pt in the Pt/Ti-PLA and Pt/Ti-P25 samples calcined at 600 °C are 16 nm and 11 nm, respectively. This is comparable to data reported in [16] for samples aged under air in presence of water. An increase in the calcination temperature leads to a change in the size of Pt crystallites (Table 1). For the support obtained by PLA, the size of Pt crystallites is slightly larger. The amount of platinum observed by XRD is lower than the actual platinum loading in the catalysts even after their calcination at 800 °C. Hence, approximately one third of the platinum in both samples remains in an X-ray amorphous or highly dispersed state.
3.2. HRTEM Study
HRTEM images of Pt/Ti-PLA-400 and Pt/Ti-P25-400 samples are shown in Figure 2 and Figure 3, respectively. The size of the support particles in the Pt/Ti-PLA-400 catalyst varies in a wide range from 5 to 75 nm. HRTEM shows that the distribution of platinum particles over a support is non-uniform (Figure 2a,b). The average size of platinum particles is 1.4 nm (Figure 2b, insert). The non-uniform particle size distribution leads to the formation of extended structures on the surface due to partial agglomeration of nearby nanoparticles (marked with yellow circles in Figure 2c). The platinum nanoparticles are often localized at the intercristalline boundaries of the support particles (Figure 2d).

Figure 2.
(a,c,d) High-resolution transmission electron microscopy (HRTEM) and (b) high-angle annular dark-field imaging scanning transmission electron microscopy (HAADF-STEM) images for Pt/Ti-PLA-400 sample. The inset shows the size distribution of platinum particles.
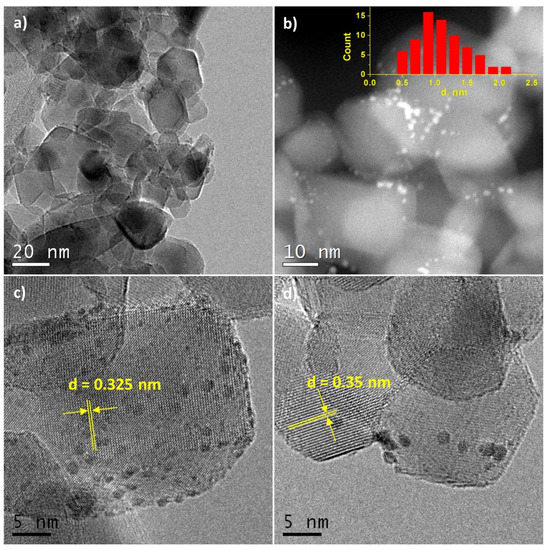
Figure 3.
(a,c,d) High-resolution transmission electron microscopy (HRTEM) and (b) high-angle annular dark-field imaging scanning transmission electron microscopy (HAADF-STEM) images for Pt/Ti-P25-400 sample. The inset shows the size distribution of platinum particles.
In the Pt/Ti-P25-400 catalyst, the support is represented by nanoparticles of 10–30 nm in size. HRTEM and HAADF-STEM images (Figure 3a,b) clearly show platinum nanoparticles distributed over the entire surface of the support. The average size of the Pt particles is 1.1 nm (Figure 3b, insert). The images uncover that Pt particles smaller than 1 nm are present in the sample. The size distribution curve as well as the contrast heterogeneity of the support lattice fringe in the high-resolution images (Figure 3c,d), also suggest the formation of highly dispersed platinum species. Interplanar distances measured from the high-resolution images of the support nanoparticles correspond to two phases of titanium dioxide: anatase (d101 = 0.352 nm) and rutile (d110 = 0.324 nm).
HRTEM and XRD data for catalysts calcined at 400 °C are in a good agreement, pointing to the formation of small Pt particles. The distribution of platinum particles over the surface of the TiO2-PLA support suggests that the platinum particles are stabilized on the TiO2-brookite or TiO2-rutile particles (identified by analysis of interplanar distances) which, according to XRD data, are smaller than TiO2-anatase particles. In the Pt/TiO2-P25-400 catalyst anatase and rutile TiO2 are present as highly crystalline particles of similar size. The preferential stabilization of platinum particles, especially particles less than 1 nm, on the surface of rutile particles has also been observed but is less pronounced (Figure 3c,d).
3.3. Raman Spectroscopic Analysis
Phase transformations of titanium oxide with increase of the calcination temperature were also analyzed by Raman spectroscopy. Figure 4 shows evolution of the Raman spectra of the Pt/Ti-PLA (Figure 4a) and Pt/Ti-P25 (Figure 4b) samples depending on the calcination temperature. The optical range 300–800 cm–1 is usually used to detect phase transformations of titanium oxide [38]. In this range, the B1g (396 cm–1), A1g + B1g (396 cm–1) and Eg (637 cm–1) modes related to TiO2 anatase and Eg (444 cm–1), A1g (609 cm–1) modes related to TiO2 rutile are located. The Pt/Ti-P25 catalyst changes significantly even after calcination at 600 °C (Figure 4b), as evidenced by the appearance of the rutile modes. For the Pt/Ti-PLA-600 sample the bands related to the rutile phase can hardly be noticed. Calcination at 800 °C in the case of both catalysts leads to a sharp increase of the intensity of the modes related to TiO2-rutile. The wide and intense lines completely mask the possible presence of the B1g, A1g + B1g and Eg modes of TiO2-anatase.
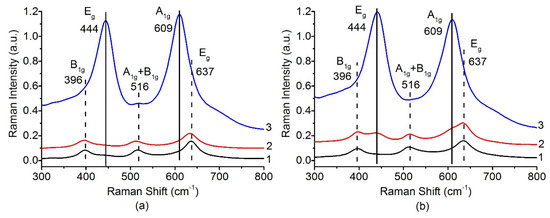
Figure 4.
Raman spectra in range 300–800 cm−1 for (a) Pt/Ti-PLA and (b) Pt/Ti-P25 catalysts calcined at 400 °C (1), 600 °C (2) and 800 °C (3).
The presence of the anatase phase can be determined by analyzing the band at 144 cm–1 corresponding to the Eg mode of the TiO2-anatase. Figure 5 shows that, in the case of a sample based on the PLA support, a significant part of TiO2 anatase phase is present even after calcination at 800 °C. For the catalyst based on the P-25 support, the anatase-to-rutile transition can be considered complete after calcination at 800 °C. These data are fully consistent with the XRD results. TiO2-PLA-based catalysts have higher thermal stability of the TiO2-anatase phase.
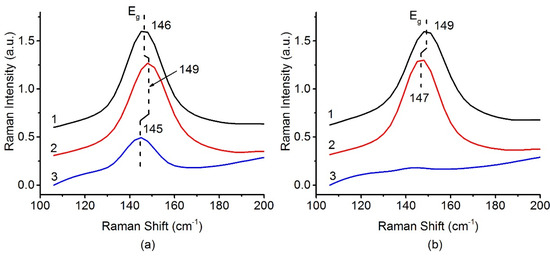
Figure 5.
Raman spectra in range 100–200 cm−1 for (a) Pt/Ti-PLA and (b) Pt/Ti-P25 catalysts, calcined at 400 °C (1), 600 °C (2) and 800 °C (3).
The small shift of the position and the change of the Eg peak width can be related to a change in the crystallite size of TiO2 anatase nanoparticles [38]. A lower frequency corresponds to an increase in the crystallite size. However, in the case of a series of Pt/Ti-PLA samples, a multidirectional shift in the line position is observed. This is in poor agreement with the XRD data and could be traced back to simultaneous growth of anatase crystallites and transformation of small brookite particles into anatase.
3.4. XANES, EXAFS
Figure 6 shows (a) XANES and (b) Fourier transformed (FT) extended X-ray absorption fine structure (EXAFS) spectra for the Pt/Ti-PLA and Pt/Ti-P25 samples. According to the XANES analysis, both samples have Pt4+ in an environment similar to PtO2, i.e., fully oxidized Pt species with oxygen atoms as nearest neighbors. In the FT EXAFS spectra, much lower contributions in the second shell than in Pt foil or PtO2 are found. To further evaluate the structure and quantify the average oxidation state of Pt sites, EXAFS spectra were fitted to several possible structural models. Statistically significant fits with realistic parameters were obtained when using Ti (Table 2) or Pt neighbors (Table A2) in the second shell; see Appendix A (Figure A1, Figure A2) for comparison of the fitted graphs in the figures. Of them, fits with Pt nearest neighbors have significant misfit at high distances and significantly different Debye–Waller factors for Pt and O shells which would be very difficult to explain for a single stable oxide compound such as PtO2. Therefore, the fits involving O and Ti shells are strongly preferred, and the resulting structural models (Table 2) suggest single oxidized Pt atoms strongly bonded to the TiO2 support.
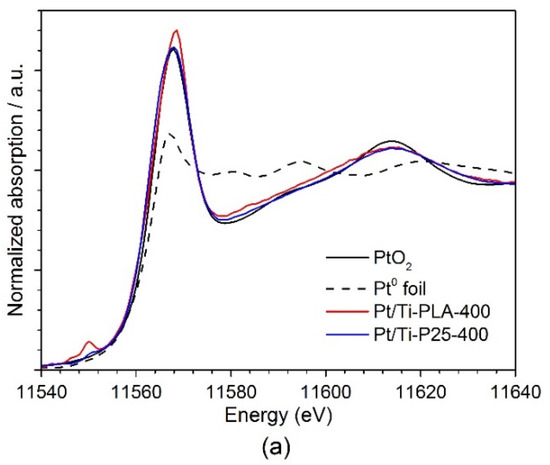
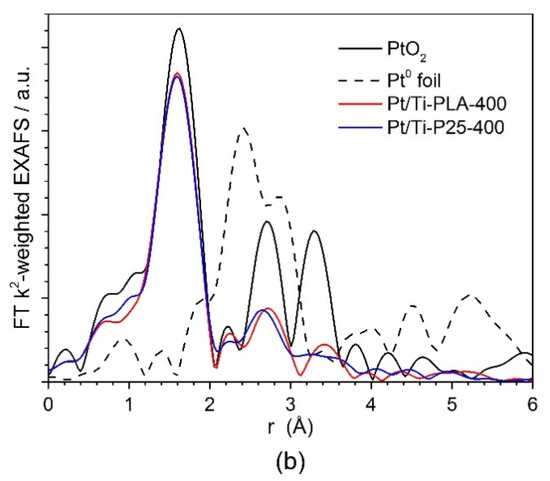
Figure 6.
(a) Ex situ XANES and (b) Fourier transformed (FT) k2-weighted EXAFS (uncorrected for the phase shift) spectra of Pt/Ti-PLA and Pt/Ti-P25 samples, and data for PtO2 and metallic Pt reference samples. The EXAFS is Fourier-transformed in the k-range of: 2.5–11.0 Å−1.
Table 2.
Results of extended X-ray absorption fine structure (EXAFS) fitting to a structural model containing O backscatterers (first shell) and Ti backscatterers (second shell) as nearest neighbors.
3.5. XPS Analysis
3.5.1. Analysis of the Ti2p and O1s Core-Level Spectra
X-ray photoelectron spectroscopy (XPS) was performed to study the charge states of the elements on the surface. According to the XPS data (Figure A3), the state of titanium in all samples is characterized by a single doublet with a binding energy value Eb(Ti2p3/2) = 458.6 eV, which, according to literature data [35], corresponds to Ti4+ in titanium dioxide. The half-width of the titanium line does not change significantly and amounts to 1.6 eV for the Ti2p3/2 component and 2.4 eV for Tip1/2. The phase transition between rutile and anatase does not lead to a change in the charge state of titanium, and no change in the position and shape of the Ti2p core line can be observed.
The spectral region of oxygen O1s contains two main lines at Eb (O1s) = 529.9 and 531.7 eV (Figure A3c,d) which correspond to oxygen in the lattice of titanium oxide and oxygen on the surface defects, respectively [29]. In case of the Pt/Ti-P25-800 sample oxygen is represented by three states. However, it should be taken into account that in this sample a large amount of carbon impurities is present on the surface (Table 3), some of which are related to the oxygen-containing COx groups. The adsorption of hydrocarbon (HC) impurities on the surfaces is well known, e.g., due to back diffusion from the pumps. In fact, this HC is often used as an internal standard for XP spectra calibration. The increase in calcination temperature does not lead to carbon burning, proving that it is not an internal part of the samples. It should be considered as inert impurities on the surface, not affecting catalytic properties. From the presented Ti2p and O1s spectra, the O/Ti atomic ratios (Table 3) were calculated with the contribution of the lattice oxygen (Olat) and the surface oxygen groups (Oads) of the OH and COx type (Eb = 531.5–531.7 eV). The Oads/Olat ratio is lower for the Pt/Ti-PLA catalysts than for the Pt/Ti-P25 catalyst series. This ratio can be considered as an indirect indication of a different number of surface defects due to a different titania synthesis route.
Table 3.
Charge states and surface concentrations (atomic %) of the elements by X-ray photoelectron spectroscopy (XPS).
3.5.2. Analysis of Pt4f and Ti3s Spectral Region
In order to obtain the correct data on the state of platinum from the analysis of the Pt4f spectra it was necessary to subtract the contribution of the lines from the TiO2 support within the Pt4f spectral region. The Ti3s line is characterized by the presence of a satellite structure/background of scattered electrons in the range of 70–80 eV with a maximum intensity in at 75.7 eV (Figure 7a). Despite the low intensity of the Ti3s line and even lower intensity of the satellite, their impact can significantly complicate analysis of the Pt4f spectra for Pt/TiO2 samples with low platinum loading. To obtain a corrected Pt4f spectrum, the background line recorded for a pure TiO2 support was subtracted from the experimental spectrum of Pt/TiO2 sample. The intensity of a given background line should be normalized by the Ti3s line recorded for the catalyst and pure support. However, the Ti3s line has low intensity and, in order to prevent surface reduction by long X-ray exposure, the line of the subtracted background was normalized to the intense of Ti2p line.
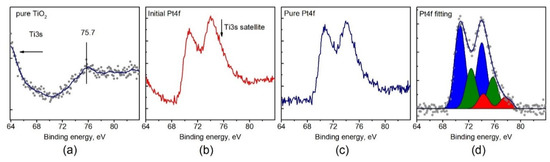
Figure 7.
(a) The XPS spectrum of Ti3s satellite for pure TiO2 support; (b) the experimental XPS spectrum of the Pt4f region for Pt/TiO2 catalyst; (c) the Pt4f spectrum after Ti3s satellite subtraction; (d) the curve-fitted Pt4f spectrum.
Figure 7b,c illustrates that the Ti3s satellite, with a maximum at 75.7 eV, can influence the platinum line shape, provided that the intensity of platinum line is low either due to low dispersion or loading of platinum. Even for relatively high loading of platinum (about 2%), the integral line shape is distorted and the Pt4f5/2 component becomes more intense than Pt4f7/2 which has no physical meaning. Thus, the presence of the Ti3s satellite line should be taken into account when analyzing Pt/TiO2 catalysts by XPS.
Using the described procedure, corrected Pt4f spectra were obtained for all samples. Concentrations of all elements on the surface with contributions of the individual oxidation states were estimated from the XPS data (Table 3). The increase of the calcination temperature leads to the decrease of the total platinum amount observed by XPS. As XPS is a surface sensitive technique, this decrease might be caused by sintering of platinum in accordance with the XRD data (Table 1). The sintering of platinum is accompanied by TiO2 particle growth. At 800 °C, the decrease in the TiO2 surface is more pronounced compared to the Pt surface. Hence, a slight increase of the Pt/Ti ratio after calcination at 800 °C is observed.
3.5.3. Analysis of Pt4f Spectra
The curve fitted Pt4f spectra are shown in Figure 8. Each doublet corresponds to the individual state of platinum on the surface of the catalysts (Table 3). For Pt/Ti-PLA-400, two states with Eb (Pt4f7/2) = 71.7 eV and 73.7 eV can be distinguished. For Pt/Ti-P25-400, two peaks with Eb (Pt4f7/2) = 72.7 and 74.9 eV can be seen. Note that the Pt4f7/2 binding energies for both platinum states in the Pt/Ti-P25-400 catalyst are 1.0–1.2 eV higher than in the Pt/Ti-PLA-400 catalyst pointing to higher oxidation state of platinum. The state with Eb = 74.9 eV can be assigned to Pt4+ [13,36], while the Pt state with Eb = 73.7 eV has a lower value than the typical values for the Pt4+ ion [22,36,39].
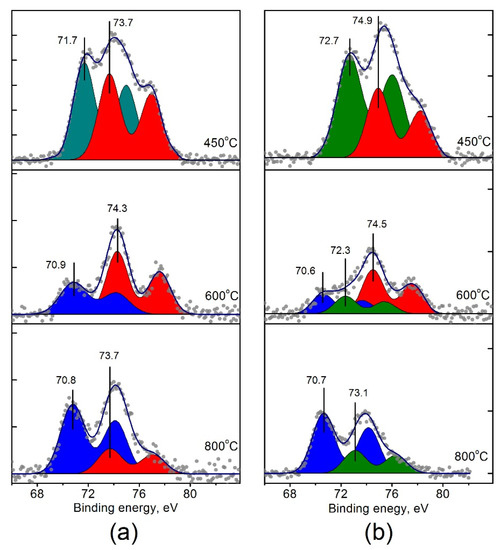
Figure 8.
Curve-fitted Pt4f XPS spectra for (a) Pt/Ti-PLA and (b) Pt/Ti-P25 catalysts, calcined at 400 °C, 600 °C and 800 °C.
The state with Eb = 72.7 eV, observed for the Pt/Ti-P25-400 catalyst, can be attributed to Pt2+ platinum ions [36,39]. As for the peak with Eb = 71.7 eV obtained for the Pt/Ti-PLA-400 catalyst, it can be related both to metal particles with a slight positive charge (Ptδ+) due to contact with the oxide support [40,41], or to oxide particles PtOx with x < 1. In any case, this state of platinum differs from the metallic Pt0 or oxide PtO states due to interaction with the support. Thus, samples calcined at 400 °C contain ionic forms of platinum on the surface. Calcination of the catalysts at T = 600 and 800 °C leads to a formation of platinum species with Eb (Pt4f7/2) = 70.6–70.9 eV (Figure 8).
3.6. Temperature-Programmed Reduction (TPR-H2)
Figure 9 presents results of TPR-H2 experiments for the Pt/Ti-PLA-400 and Pt/Ti-P25-400 samples. The data on H2 consumption and H2O evolution are provided. Both H2 consumption and H2O evolution curves reveal significant differences for the analyzed catalysts. For the Pt/Ti-P25-400 catalyst, two hydrogen consumption peaks are observed with maxima at –2 and 24 °C (Figure 9b). Such low consumption temperatures are typical for PtOx clusters and oxidized platinum nanoparticles [42]. For the Pt/Ti-PLA-400 catalyst, only one sharp peak with a maximum at 4 °C can be seen (Figure 9a).
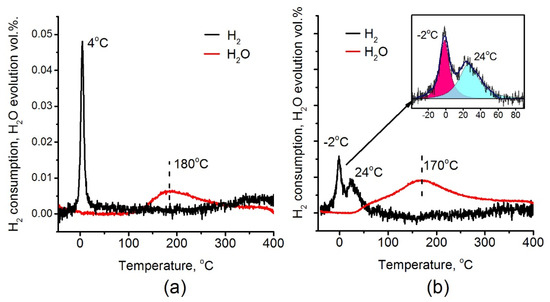
Figure 9.
H2 consumption and H2O evolution during temperature-programmed reduction (TPR-H2) for (a) Pt/Ti-PLA-400 and (b) Pt/Ti-P-25-400 catalysts.
3.7. Catalytic Activity Measurements
Figure 10 shows temperature dependences of NH3 conversion (a) and concentrations of N2 (b), N2O (c) and NOx (d) during the temperature- programmed reaction of NH3 with O2 (TPR-NH3+O2) up to 400 °C. The results obtained during two consecutive heating/cooling cycles are presented. As can be seen from the conversion curves in Figure 10a, during the first heating the catalyst based on the P25 support is the most active with the onset temperature ~120 °C. However, during the second heating, the catalytic activity decreased and the profile becomes similar to ones of the Pt/Ti-PLA-400 catalyst. The Pt/Ti-PLA-400 sample exhibits high stability and little changes from the first to second cycle of TPR-NH3+O2.
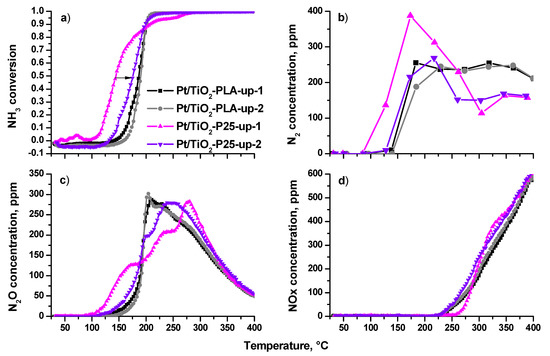
Figure 10.
(a) NH3 conversion; (b) N2 concentration, (c) N2O concentration and (d) NOx concentration in reactor as function of the temperature for Pt/Ti-PLA-400 and Pt/Ti-P25-400 catalysts in two catalytic cycles (up-1—first sample heating in the reaction medium; up-2—the second heating in the reaction medium).
The high activity of the Pt/Ti-P25-400 catalyst in the first cycle is accompanied by a higher selectivity to N2 (Figure 10b), but, after heating of the catalyst to T ~ 200 °C, the N2 concentration decreases and reaches a minimum level for all catalysts. During the second heating, N2 concentrations over Pt/Ti-P25-400 and Pt/Ti-PLA-400 are similar up to 230 °C. The N2 concentration for Pt/Ti-P25-400 catalyst drops above 230 °C, while it stays almost constant for Pt/Ti-PLA-400 catalyst.
The difference between the catalysts is more pronounced if one analyzes temperature dependences of N2O concentration. For the Pt/Ti-P25-400 catalyst, the first cycle of TPR-NH3+O2 is characterized by a three-stage growth of N2O concentration with a shift of the maximum N2O production towards higher temperatures. During the second heating the low-temperature (100–150 °C) activity disappears.
Figure 11 shows the conversion curves as a function of temperature for the catalysts calcined at T = 400 °C and 600 °C (N2 selectivity is presented in Table A3). For the Pt/Ti-P25 catalyst, an increase in the calcination temperature from 400 to 600 °C leads to a significant decrease of the low-temperature activity and T50 increases to 185 °C. In contrast, for the Pt/Ti-PLA catalyst, calcination at 600 °C leads to an increase of the catalytic activity, and T50 decreases to ~ 180 °C. The temperature of 100% conversion also significantly decreases. A comparison of the light-off curves for the catalysts calcined at 600 °C shows that the catalyst prepared with TiOx-PLA becomes more active compared to the catalyst based on the commercial P25 support.
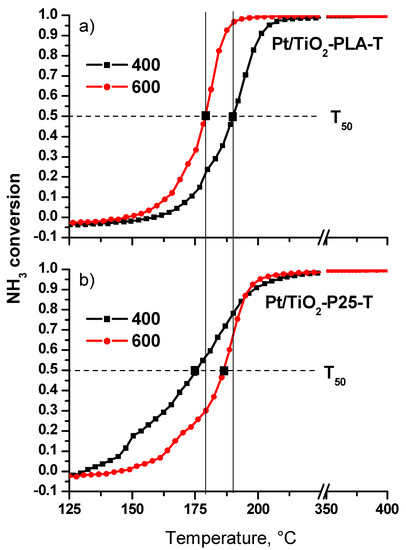
Figure 11.
NH3 conversion curves for the first catalytic cycle for the (a) Pt/Ti-PLA and (b) Pt/Ti-P25 catalysts calcined at 400oC and 600oC.
Figure A4 shows the N2 and N2O selectivity depending on the temperature of the NH3+O2 reaction. The profiles differ significantly for the Pt/Ti-PLA-400 (a, c) and Pt/Ti-P25-400 (b, d) catalysts. Thus, a characteristic feature for the Pt/Ti-PLA-400 catalyst is a sharp transition from a high N2 selectivity level (70%) to a level of 50–55%, which is almost constant over a wide temperature range from 200 to 350 °C. The switch of the reaction mode is highlighted by blue color in the temperature range 180–210 °C. This transition is observed both during the first and second heating of the catalyst, which indicates the complete reversibility catalysts activity.
Changes in the concentrations of the N2 and N2O reaction products can reflect the surface reactions of adsorbed species occurring on active centers acting in the different temperature ranges. However, the differences between the active sites are not visible at temperatures above 250 °C NO and NO2 start to form, the concentrations of which are comparable for all catalysts during the first and second heating.
The N2 and N2O selectivities observed over the Pt/T-P25-400 catalyst follow different temperature trends (Figure A4b,d). The main difference is that the transition from a highly selective reaction to N2 (70%) to low selectivity (40%) occurs in a wide temperature range in two steps. For the first heating of the catalyst, the reaction mode transition is observed in the temperature range 200–300 °C with a selectivity change from 70% to 60%. The second heating shifts the transition region by ~ 50 °C towards lower temperatures when the selectivity to N2 drops to ~ 40%. Thus, the Pt/TiO2 catalyst based on the commercial P25 support is significantly less stable than the catalyst prepared using the TiO2-PLA, which shows much higher stability and higher selectivity to N2 over a wide temperature range.
4. Discussion
The laser ablation of the Ti target in liquid followed by calcination of the resulted dispersion produces a mixture of rutile, anatase and brookite TiO2 phases. According to XRD data, the anatase and rutile particles are smaller than the one of brookite. A large amount of intercristalline boundaries “rutile–anatase” and “brookite–anatase” could be a key factor for the enhanced thermal stability of TiO2-anatase particles. The anatase phase could be observed even after calcination at 800 °C. It could be seen from TEM data that platinum deposition is not uniform and preferentially takes place on the intercristalline boundaries or the surface of the anatase particles. Thus, microheterogeneity of TiO2 produced by PLA can be responsible for the increase in the thermal stability of TiO2 support as well as stabilization of the small Pt particles.
According to the XPS data, platinum in the catalysts calcined at 400 °C is in the ionic form, regardless of the type of the TiO2 support. Metal clusters or metallic Pt nanoparticles are not observed. The highly oxidized forms of platinum in the catalysts have different binding energies: 73.7 eV for Pt/Ti-PLA-400 and 74.9 eV for Pt/Ti-P25-400. A significant difference in the binding energy –1.2 eV can be explained by different sizes of Pt particles and different degrees of interaction with the support. Highly dispersed platinum particles of less than 1 nm size are present on the surface of the commercial TiO2-P25 support, while the TiO2-PLA support stabilizes particles larger than 1 nm. Probably due to the small size of platinum particles on the surface of the TiO2 – P25 support, the Pt particles are completely oxidized to Pt4+ species [22].
For larger particles on the PLA support a partial reduction of Pt ions might be proposed. In this case, splitting of the Pt4f line into the two doublets from Pt2+ and Pt4+ forms would be expected. However, in the Pt4f spectra of the Pt/Ti-PLA-400 sample we observed only one doublet from PtOx species. The Eb (Pt4f7/2) = 73.7 eV, determined from the Pt4f spectra for this state, is the intermediate value between the one typical for Pt2+ and Pt4+ ions. In the literature, this state was even described as a Pt3+ state [43]. At the same time Pt3O4 is described as a mixed oxide with defined Pt4+ and Pt2+ sites in the structure [44]. Thus we tentatively associate the Pt state in the Pt/Ti-PLA-400 sample with slightly reduced PtO2 particles.
Species with Eb (Pt4f7/2) = 70.6–70.9 eV observed after catalyst calcination at T = 600 and 800 °C species (Figure 8) can be attributed to highly dispersed metallic platinum particles with a negative charge (Ptδ−) due to electronic interaction with TiO2. In this case, Pt4f binding energy is affected by photoemission relaxation effects [45,46]. According to literature data [47,48], for small Pt particles (less than 1 nm) the shifts of Pt4f7/2 line relative to the Eb value of bulk Pt can reach 0.6 eV. Taking the value of Eb (Pt4f7/2) for the bulk metal as 71.1 eV [49], the obtained experimental values of 70.6–70.9 eV correspond to −0.5 ÷ −0.2 eV shifts. The slightly lower values of Eb (Pt4f7/2)= 70.6–70.7 eV and, accordingly, the larger shifts of the Pt4f doublet towards lower Eb observed for Pt/Ti-P25 catalysts can point to the presence of smaller Pt particles in these catalysts. This is also in a good agreement with the HRTEM data.
Despite the common trend in the transition of ionic forms of platinum to metal with an increase in the calcination temperature of the catalysts, the resulting Pt4f spectra show some differences. For the Pt/Ti-PLA-600 catalyst only two forms of platinum can be observed: Ptδ− (Eb = 70.9 eV) and Pt4+ (Eb = 74.3 eV). At the same time, in the Pt/Ti-P25-600 catalyst three platinum forms are present: Ptδ− (Eb = 70.6 eV), Pt2+ (Eb = 72.3 eV) and Pt4+ (Eb = 74.5 eV). A highly oxidized form of platinum with Eb = 73.7 eV is present in the TiO2-PLA-based catalyst even after calcination at 800 °C, demonstrating higher thermal stability than in the case of the Pt/Ti-P25 catalysts. The increased stability of the ionic forms of platinum might be associated with their localization at the boundaries of TiO2 particles in the Pt/Ti-PLA catalyst. According to the XRD and HRTEM data, small particles of TiO2-anatase and TiO2-brookite cover the TiO2-rutile. In this case, oxidized platinum is in contact with anatase and brookite surfaces.
According to XPS, platinum in the Pt/Ti-PLA-400 catalyst has lower binding energies indicating lower stoichiometry of oxygen in PtOx particles. The quantitative estimation of the Pt/O ratio obtained from TPR-H2 data are in good agreement with the XPS data. Fitting of the hydrogen consumption curve for the Pt/Ti-P25-400 sample (Figure 9b, inset) shows that the areas of the individual consumption peaks are very similar. The XPS data give the ratio of the Pt2+: Pt4+ components as 2:1. Hence, the amount of hydrogen consumed by the PtO oxide should be similar to the one for the PtO2 oxide in good agreement with the TPR-H2 data.
The two hydrogen consumption peaks observed for the Pt/Ti-P25-400 catalyst indicate two nonequivalent states of oxygen either as a part of one PtOx structure or as separate PtO2 and PtO clusters. The low-temperature peak has a maximum at T = −2 °C. This is a lower temperature value than was observed for the Pt/Ti-PLA-400 catalyst. It indicates higher reactivity of oxygen in the Pt/Ti-P25-400 catalyst. In addition, the total amount of consumed hydrogen is slightly higher for the Pt/Ti-P25-400 catalyst (150 μmol/g versus 128 μmol/g for Pt/Ti-PLA-400 catalyst) which gives the H2/Pt ratio of 1.46 and 1.28, respectively.
A comparison of the H2O evolution curves shows that, in the case of the Pt/Ti-PLA-400 catalyst, the evolution occurs at temperatures above 120 °C as a single peak with a maximum at T = 180 °C. H2O evolution for the Pt/Ti-P25-400 catalyst occurs in two stages, one of which is described by a low-temperature peak with maximum at T~ 60 °C. The appearance of a low-temperature peak indicates formation of the weakly-bonded reactive OH groups on the surface of the Pt/Ti-P25-400 catalyst. As a consequence, in the case of the Pt/Ti-P25-400 catalyst, the H2O evolution takes place along with the second hydrogen consumption peak. While for the Pt/Ti-PLA-400 catalyst the significant temperature difference (4–120 °C) between the consumption of H2 and the evolution of water is observed due to formation of more strongly bound OH groups.
For the Pt/Ti-PLA-400 catalyst the single peak in the TPR-H2 (Tmax= 4 °C) has narrow width and high intensity. These facts indicate a high homogeneity of the reactive oxygen species. It can probably be associated with highly oxidized Pt with Eb (Pt4f7/2) = 73.7 eV. The XPS spectra also show a platinum component with the binding energy Eb (Pt4f7/2) = 71.7 eV (Figure 8a), designated as Ptδ+. This state is formed due to the strong interaction of Pt with the TiO2 surface. It could be described as PtnOm clusters, where m < n and the platinum state is intermediate between the Pt0 and Pt2+. Note that these structures do not give a separate peak on the hydrogen consumption curve in TPR-H2 experiments but result in the broadened peak in the region up to 200 °C. However, oxygen in these structures affects the charge state of platinum. The formation of Pt-O-Ti bonds can be proposed in good agreement with results of EXAFS analysis.
The decrease of catalytic activity after annealing at high temperatures is a common effect. In our case it could be explained by sintering of platinum nanoparticles, leading to decreased amount of active sites. In case of TiO2 supports platinum surface could be passivated by migration of TiO2 on the Pt particles (SMSI effect). For samples studied in the present work annealing at 600 and 800 °C leads to TiO2 phase transition from anatase to rutile. This process is accompanied by the decrease of surface area of the support which, in turn, leads to reduction and growth of Pt particles. In the case of the Pt/TiO2-P25 catalyst, platinum particles are weakly bonded to the surface. During heating under reaction conditions, platinum easily reduces and sinters, leading to the decrease of catalytic activity. In the case of the Pt/TiO2-PLA sample, these processes are less pronounced due to stronger interaction between platinum and TiO2. As it was shown by XPS and TEM, platinum particles preferentially stabilize on rutile surfaces. As TiO2 rutile is more stable in the PLA support, reduction and sintering of platinum are slowed down and the higher catalytic activity is maintained.
Previously it was shown that in the course of the catalytic reaction of NH3 with O2 a transition from the oxidized to partially reduced platinum species takes place [30,50]. TPR-H2 data shows that the reduction processes of Pt/Ti-P25-400 catalysts and Pt/Ti-PLA-400 are different. Therefore, it is of interest to compare the catalysts in their reduced state after the catalytic cycle, i.e., after exposure to the reaction medium. The XPS spectra of the Pt/Ti-PLA-400 and Pt/Ti-P25-400 catalysts before and after their exposure to the reaction medium were compared. Figure 12 shows Pt4f spectra collected for the samples after the first TPR-NH3+O2 cycle. The spectra reveal that, despite the higher degree of oxidation of platinum in the initial Pt/Ti-P25-400 catalyst, the degree of reduction of this catalyst in the reaction medium is much stronger. The fraction of the ionic form of platinum with Eb (Pt4f7/2) = 72.4–72.7 eV (Pt2+) becomes lower (Table 4), and Pt4+ ions with Eb (Pt4f7/2) = 74.3 eV completely disappear.
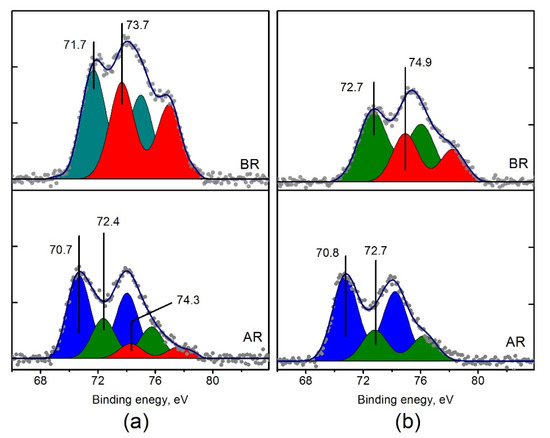
Figure 12.
Curve fitted Pt4f spectra of (a) Pt/Ti-PLA-400 and (b) Pt/Ti-P25-400 catalysts before (BR) and after their exposure to reaction medium (AR). The spectra of the catalysts before catalytic experiments are given to allow comparison with the spectra obtained after the catalytic cycle.
Table 4.
Relative distributions of platinum forms in the catalysts (as a percentage to the total amount of platinum) in the samples before and after exposure to the reaction medium.
The Pt4f spectra of the Pt/Ti-PLA-400 and Pt/Ti-P25-400 catalysts after exposure to the reaction atmosphere show higher degree of similarity and consist of two main doublets with Eb (Pt4f7/2) = 70.7–70.8 eV (Ptδ−) and 72.4–72.7 eV (Pt2+). However, the ratio between these platinum species is different. Pt2+/Ptδ− ratio is 0.5 for the Pt/Ti-PLA-400 catalyst, while it is 0.4 for the Pt/Ti-P25-400 catalyst (Table 4).
Based on the characterization data, the higher activity of the Pt/Ti-P25-400 catalyst during the first catalytic cycle can be explained as follows. The Pt/Ti-P25-400 catalyst contains highly dispersed clusters with a high Pt oxidation state. These clusters are characterized by two types of oxygen with reduction temperatures of −2 and 24 °C;. In the Pt/Ti-PLA-400 catalyst, there is only one type of oxygen with the reduction temperature of 4 °C. Furthermore, the evolution of water during the reduction is observed at different temperatures. For the Pt/Ti-PLA-400 catalyst, water evolution takes place above 120 °C. For the Pt/Ti-P25-400 catalyst H2O evolution begins at about 24 °C, exactly in the temperature range in which the peak of hydrogen consumption is observed. We can conclude that more strongly bonded -OH groups are formed on the Pt/Ti-PLA-400 catalyst, which prevent adsorption of NH3 and O2 and, as a result, the NH3 dehydrogenation reaction [51]. On the Pt/Ti-P25-400 catalyst, along with the strongly bonded OH groups, the weakly bonded OH groups are formed which recombine and desorb from the surface at a significantly lower temperature. This provides sites for the adsorption of the reactants. After the first heating of the catalysts in the reaction mixture, the state of Pt in both catalysts is almost the same, which results in a similar activity during the second heating.
5. Conclusions
This work presents a comprehensive study by physicochemical methods, including X-ray photoelectron spectroscopy, of the Pt/TiO2 catalysts prepared on the basis of a standard commercial TiO2-P25 support and on a TiO2 support synthesized using a pulsed laser ablation in liquid (PLA). The TiO2 support synthesized by the PLA method contains rutile, brookite, and anatase phases with different particle sizes. Smaller particles of the rutile and brookite phases are distributed over the surface of the larger particles of anatase TiO2. The Pt/TiO2-PLA catalyst obtained by impregnation of the PLA-support showed enhanced thermal stability compared to the catalyst based on the commercial P25 support. After calcination of the Pt/TiO2-PLA catalyst at 800 °C, the anatase phase is unambiguously detected by X-ray diffraction and Raman spectroscopy in contrast to the Pt/TiO2-P25 catalyst, in which the anatase phase completely transforms to rutile. HRTEM shows that the Pt/TiO2-PLA-400 catalyst contains platinum particles with an average size of 1.4 nm. The distribution of Pt particles over the surface of the support is not uniform. Particles preferentially localize at the interblock boundaries of the support and partially agglomerate. Larger particles are fixed mainly on the rutile surface, in contrast to the Pt/TiO2-P25 sample, in which platinum is uniformly distributed over the entire surface of the support.
The states of the components of the catalysts were analyzed using the XPS after calcination in air at various temperatures and after exposure to the reaction medium. Annealing of the conventional Pt/TiO2-P25 catalyst at 400 °C leads to the formation of oxidized platinum structures PtO and PtO2. In the Pt/TiO2-PLA-400 catalyst the oxidation state of platinum is lower due to the stronger interaction of the PtOx clusters (x < 1) with the support. The formation of Pt-O-Ti structures is confirmed by EXAFS data. This fact is also confirmed by TPR-H2 experiments, which showed the presence of a single reactive state of oxygen in the Pt/TiO2-PLA-400 sample, in contrast to the two forms of oxygen in the Pt/TiO2-P25-400 catalyst. The Pt4f spectra of the samples after reaction show the residual form of Pt2+ and the metallic form of platinum with a negative shift of the Pt4f doublet relative to the value typical for the bulk Pt metal. It was interpreted as the formation of Ptδ− states. Calcination at 600 and 800 °C resulted in decomposition of the oxidic Pt structures and sintering of the support particles. These processes are more pronounced for the catalyst prepared with the commercial P25 support, apparently due to the sintering and recrystallization of anatase particles.
The catalytic properties of the samples were studied in the ammonia oxidation reaction. The Pt/Ti-P25-400 catalyst demonstrates higher NH3 conversion at low temperatures in the first catalytic NH3+O2 cycle, however, the subsequent cycle suppresses this low-temperature activity. Unlike the conventional Pt/Ti-P25 catalyst, the Pt/Ti-PLA catalyst shows high stability in all cycles of the temperature-programmed reaction, with a significantly higher selectivity for the N2 product in a wide temperature range of 200–400 °C.
Thus, it has been shown that the use of the pulsed laser ablation for the synthesis of the TiO2 support can provide significant advantages in comparison with the standard TiO2-P25 support. These include increased thermal stability of the crystalline phases of the support and the lower charge state of active component due to the stronger interaction with the support. All these factors play an important role for the practical application of ammonia slip catalysts.
Author Contributions
XRD analysis, E.F., HR TEM analysis, O.S., XAS analysis D.D., V.M., Catalytic measurements, E.S., D.S., PLA synthesis, I.L., E.F., PLA synthesis and Raman analysis, V.S., Catalysts synthesis, A.R., writing—original draft preparation and XPS analysis, A.S., writing—review and editing, L.K., E.S., A.B., J.-D.G., discussion, supervision and project administration, A.B., J.-D.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Helmholtz–Russian Science Foundation Joint Research Groups grant #18-43-06201 from 03.09.2018 (RSF)/HRSF-0046 from 01.09.2018 (HGF).
Acknowledgments
We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provided beam time. Parts of this research were carried out at PETRA III and we would like to thank Edmund Welter for assistance in using beamline P65. We further thank Dr. Paolo Dolcet (KIT) for his support during XAS measurements.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Appendix A

Figure A1.
(a) Pt/Ti-P25-400 EXAFS fit to a structural model containing O and Ti nearest neighbors; (b) Pt/Ti-PLA-400 EXAFS fit to a structural model containing O and Ti nearest neighbors.
Figure A1.
(a) Pt/Ti-P25-400 EXAFS fit to a structural model containing O and Ti nearest neighbors; (b) Pt/Ti-PLA-400 EXAFS fit to a structural model containing O and Ti nearest neighbors.


Figure A2.
(a) Pt/Ti-P25-400 EXAFS fit to a structural model containing O and Pt nearest neighbors; (b) Pt/Ti-PLA-400 EXAFS fit to a structural model containing O and Pt nearest neighbors.
Figure A2.
(a) Pt/Ti-P25-400 EXAFS fit to a structural model containing O and Pt nearest neighbors; (b) Pt/Ti-PLA-400 EXAFS fit to a structural model containing O and Pt nearest neighbors.


Figure A3.
Ti2p fitted XPS spectra of (a) Pt/Ti-PLA-400 and (b) Pt/Ti-P25-400 catalysts; O1s fitted XPS spectra of (c) Pt/Ti-PLA-400 and (d) Pt/Ti-P25-400 catalysts, calcined at 400 °C (1), 600 °C (2) and 800 °C (3). O1s spectra are normalized to the Ti2p area of Pt/Ti-PLA-400 sample.
Figure A3.
Ti2p fitted XPS spectra of (a) Pt/Ti-PLA-400 and (b) Pt/Ti-P25-400 catalysts; O1s fitted XPS spectra of (c) Pt/Ti-PLA-400 and (d) Pt/Ti-P25-400 catalysts, calcined at 400 °C (1), 600 °C (2) and 800 °C (3). O1s spectra are normalized to the Ti2p area of Pt/Ti-PLA-400 sample.


Figure A4.
Selectivity for N2 and N2O products depending on the NH3 + O2 reaction temperature during two heating cycles for the catalysts: (a) Pt/Ti-PLA-400; (b) Pt/Ti-P25-400. N2O selectivity was calculated based on the experimental measurements, N2 selectivity was calculated by balance. The transition region from a high N2 selectivity level is filled with blue; the region of low N2 selectivity is marked with green color.
Figure A4.
Selectivity for N2 and N2O products depending on the NH3 + O2 reaction temperature during two heating cycles for the catalysts: (a) Pt/Ti-PLA-400; (b) Pt/Ti-P25-400. N2O selectivity was calculated based on the experimental measurements, N2 selectivity was calculated by balance. The transition region from a high N2 selectivity level is filled with blue; the region of low N2 selectivity is marked with green color.

Table A1.
Structural characteristics of TiO2 in Pt/Ti-PLA and Pt/Ti-P25 samples.
Table A1.
Structural characteristics of TiO2 in Pt/Ti-PLA and Pt/Ti-P25 samples.
| Sample | TiO2-Anatase | TiO2-Rutile | TiO2-Brookite | Pt | ||||
|---|---|---|---|---|---|---|---|---|
| a, Å | c, Å | a, Å | c, Å | a, Å | b, Å | c, Å | a, Å | |
| Pt/Ti-PLA 400 | 3.785 | 9.511 | 4.593 | 2.959 | 9.19 | 5.438 | 5.143 | - |
| Pt/Ti-PLA 600 | 3.785 | 9.512 | 4.593 | 2.960 | 9.19 | 5.438 | 5.143 | 3.922 |
| Pt/Ti-PLA 800 | 3.784 | 9.515 | 4.593 | 2.959 | - | - | - | 3.923 |
| Pt/Ti-P25-400 | 3.786 | 9.508 | 4.595 | 2.960 | - | - | - | - |
| Pt/Ti-P25-600 | 3.786 | 9.512 | 4.594 | 2.962 | - | - | - | 3.922 |
| Pt/Ti-P25-800 | - | - | 4.593 | 2.960 | - | - | - | 3.923 |
Table A2.
Results of EXAFS fitting to a structural model containing O and Pt nearest neighbors.
Table A2.
Results of EXAFS fitting to a structural model containing O and Pt nearest neighbors.
| Catalyst | Pt-O | DW Factor (10−3 Å2) | Pt-Pt | DW Factor (10−3 Å2) | δE0 (eV) | ρ (%) | ||
|---|---|---|---|---|---|---|---|---|
| Distance (Å) | CN | Distance (Å) | CN | |||||
| Pt/Ti-P25 400 | 2.00 ± 0.01 | 6.0 ± 0.6 | 3.9 ± 1.6 | 3.14 ± 0.05 | 9.1 ± 6.3 | 12.2 ± 8.1 | 12.4 ± 1.0 | 0.5 |
| Pt/Ti-PLA 400 | 2.00 ± 0.01 | 5.3 ± 0.6 | 1.6 ± 2.0 | 3.14 ± 0.05 | 4.9 ± 4.0 | 3.7 ± 8.3 | 12.4 ± 2.1 | 1.4 |
Table A3.
N2 selectivity at different temperatures for catalysts calcined 400 and 600 °C.
Table A3.
N2 selectivity at different temperatures for catalysts calcined 400 and 600 °C.
| Reaction Temperature, °C | Pt/TiO2-PLA | Pt/TiO2-P25 | ||
|---|---|---|---|---|
| 400 °C | 600 °C | 400 °C | 600 °C | |
| 170–180 | 70.8 | 80.1 | 76.1 | 68.6 |
| 215–225 | 52.4 | 55.8 | 54.0 | 61.8 |
| 260–270 | 52.7 | 41.5 | 32.6 | 40.5 |
| 305–315 | 50.1 | 42.8 | 30.9 | 34.4 |
| 345–355 | 48.7 | 46.1 | 33.9 | 34.1 |
| 385–395 | 41.0 | 45.6 | 31.3 | 32.4 |
References
- Chen, M.S.; Cai, Y.; Yan, Z.; Gath, K.K.; Axnanda, S.; Goodman, D.W. Highly Active Surfaces for CO Oxidation on Rh, Pd, and Pt. Surf. Sci. 2007, 601, 5326–5331. [Google Scholar] [CrossRef]
- Oi-uchisawa, J.; Wang, S.; Nanba, T.; Ohi, A.; Obuchi, A. Improvement of Pt Catalyst for Soot Oxidation Using Mixed Oxide as a Support. Appl. Catal. B Environ. 2003, 44, 207–215. [Google Scholar] [CrossRef]
- Chen, W.; Yamada, J.; Matsumoto, K. Catalytic olefin epoxidation with molecular oxygen over supported amidate- bridged platinum blue complexes. Synth. Commun. 2002, 32, 17–22. [Google Scholar] [CrossRef]
- Chakraborty, D.; Petersen, H.N.; Elkjær, C.; Cagulada, A.; Johannessen, T. Solid Ammonia as Energy Carrier: Current Status and Future Prospects. Fuel Cells Bull. 2009, 12–15. [Google Scholar] [CrossRef]
- Son, I.H.; Shamsuzzoha, M.; Lane, A.M. Promotion of Pt/γ-Al2O3 by New Pretreatment for Low-Temperature Preferential Oxidation of CO in H2 for PEM Fuel Cells. J. Catal. 2002, 210, 460–465. [Google Scholar] [CrossRef]
- Svintsitskiy, D.A.; Kibis, L.S.; Stadnichenko, A.I. Insight into the Nature of Active Species of Pt/Al2O3 Catalysts for Low Temperature NH3 Oxidation. ChemCatChem 2019, 11, 1–15. [Google Scholar] [CrossRef]
- Yu, X.; Ye, S. Recent Advances in Activity and Durability Enhancement of Pt/C Catalytic Cathode in PEMFC Part I. Physico-Chemical and Electronic Interaction between Pt and Carbon Support, and Activity Enhancement of Pt/C Catalyst. J. Power Sources 2007, 172, 133–144. [Google Scholar] [CrossRef]
- Wang, H.; Yang, W.; Tian, P.; Zhou, J.; Tang, R.; Wu, S. Applied Catalysis A: General A Highly Active and Anti-Coking Pd-Pt/SiO2 Catalyst for Catalytic Combustion of Toluene at Low Temperature. Appl. Catal. A Gen. 2017, 529, 60–67. [Google Scholar] [CrossRef]
- Hansen, T.K. Development of New Diesel Oxidation and NH3 Slip Catalysts. Ph.D. Thesis, Technical University of Denmark (DTU), Lyngby, Denmark, 2017. [Google Scholar]
- Bruix, A.; Lykhach, Y.; Matolínová, I.; Neitzel, A.; Skála, T.; Tsud, N.; Vorokhta, M.; Stetsovych, V.; Ševčíková, K.; Mysliveček, J.; et al. Maximum Noble-Metal Efficiency in Catalytic Materials: Atomically Dispersed Surface Platinum. Angew. Chem. Int. Ed. Engl. 2014, 53, 10525–10530. [Google Scholar] [CrossRef]
- An, N.; Yu, Q.; Liu, G.; Li, S.; Jia, M.; Zhang, W. Complete Oxidation of Formaldehyde at Ambient Temperature over Supported Pt/Fe2O3 Catalysts Prepared by Colloid-Deposition Method. J. Hazard. Mater. 2011, 186, 1392–1397. [Google Scholar] [CrossRef]
- Li, F.B.; Li, X.Z. The Enhancement of Photodegradation Efficiency Using Pt–TiO2 Catalyst. Chemosphere 2002, 48, 1103–1111. [Google Scholar] [CrossRef]
- Derevyannikova, E.A.; Kardash, T.Y.; Stadnichenko, A.I.; Stonkus, O.A.; Slavinskaya, E.M.; Svetlichnyi, V.A.; Boronin, A.I. Structural Insight into Strong Pt–CeO2 Interaction: From Single Pt Atoms to PtOx Clusters. J. Phys. Chem. C 2019, 123, 1320–1334. [Google Scholar] [CrossRef]
- Dvorak, F.; Camellone, M.F.; Tovt, A.; Tran, N.; Negreiros, F.R.; Vorokhta, M.; Skala, T.; Matolinova, I.; Myslivecek, J.; Matolin, V.; et al. Creating Single-Atom Pt-Ceria Catalysts by Surface Step Decoration. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Kunwar, D.; Zhou, S.; Delariva, A.; Peterson, E.J.; Xiong, H.; Purdy, S.C.; Veen, R.; Brongersma, H.H.; Pereira-Herna, X.I.; Miller, T.; et al. Stabilizing High Metal Loadings of Thermally Stable Platinum Single Atoms on an Industrial Catalyst Support. ACS Catal. 2019, 9, 3978–3990. [Google Scholar] [CrossRef]
- Boubnov, A.; Dahl, S.; Johnson, E.; Molina, A.P.; Simonsen, S.B.; Cano, F.M.; Helveg, S.; Lemus-Yegres, L.J.; Grunwaldt, J.-D. Structure-Activity Relationships of Pt/Al2O3 Catalysts for CO and NO Oxidation at Diesel Exhaust Conditions. Appl. Catal. B Environ. 2012, 126, 315–325. [Google Scholar] [CrossRef]
- Stakheev, A.Y.; Bokarev, D.A.; Prosvirin, I.P.; Bukhtiyarov, V.I. Particle-Size Effect in Catalytic Oxidation Over Pt. Nanoparticles; Sadykov, V.A., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2019. [Google Scholar] [CrossRef]
- Hsieh, B.; Tsai, M.; Pan, C.; Su, W.; Rick, J.; Chou, H.; Lee, J.; Hwang, B. Electrochimica Acta Tuning Metal Support Interactions Enhances the Activity and Durability of TiO2-Supported Pt Nanocatalysts. Electrochim. Acta 2017, 224, 452–459. [Google Scholar] [CrossRef]
- Lee, J.; Song, I.; Heui, D. Suppressed Strong Metal—Support Interactions in Platinum on Sulfated Titania and Their Influence on the Oxidation of Carbon Monoxide. ChemCatChem 2018, 10, 1258–1262. [Google Scholar] [CrossRef]
- Wang, D.; Huang, J.; Liu, F.; Xu, X.; Fang, X.; Liu, J. Rutile RuO2 Dispersion on Rutile and Anatase TiO2 Supports: The Effects of Support Crystalline Phase Structure on the Dispersion Behaviors of the Supported Metal Oxides. Catal. Today 2020, 339, 220–232. [Google Scholar] [CrossRef]
- Hegde, M.S.; Madras, G.; Patil, K.C. Noble Metal Ionic Catalysts. Acc. Chem. Res. 2009, 42, 704–712. [Google Scholar] [CrossRef]
- Mukri, B.D.; Waghmare, U.V.; Hegde, M.S. Platinum Ion-Doped TiO2: High Catalytic Activity of Pt2+ with Oxide Ion Vacancy in Ti4+1−xPt2+xO2−x Compared to Pt4+ without Oxide Ion Vacancy in Ti4+1−xPt4+xO2. Chem. Mater. 2013, 25, 3822–3833. [Google Scholar] [CrossRef]
- Teoh, W.Y.; Mädler, L.; Beydoun, D.; Pratsinis, S.E.; Amal, R. Direct (One-Step) Synthesis of TiO2 and Pt/TiO2 Nanoparticles for Photocatalytic Mineralisation of Sucrose. Chem. Eng. Sci. 2005, 60, 5852–5861. [Google Scholar] [CrossRef]
- Strobel, R.; Baiker, A.; Pratsinis, S.E. Aerosol Flame Synthesis of Catalysts. Adv. Powder Technol. 2006, 17, 457–480. [Google Scholar] [CrossRef]
- Bera, P.; Hegde, M.S. Noble Metal Ions in CeO2 and TiO2: Synthesis, Structure and Catalytic Properties. RSC Adv. 2015, 5, 94949–94979. [Google Scholar] [CrossRef]
- Kelly, P.J.; Hisek, J.; Zhou, Y.; Pilkington, R.D.; Arnell, R.D. Advanced Coatings through Pulsed Magnetron Sputtering. Surf. Eng. 2004, 20, 157–162. [Google Scholar] [CrossRef]
- Moret, M.P.; Zallen, R.; Vijay, D.P.; Desu, S.B. Brookite-Rich Titania Films Made by Pulsed Laser Deposition. Thin Solid Films 2000, 366, 8–10. [Google Scholar] [CrossRef]
- Liu, P.; Cai, W.; Fang, M.; Li, Z.; Zeng, H.; Hu, J.; Luo, X.; Jing, W. Room Temperature Synthesized Rutile TiO 2 Nanoparticles Induced by Laser Ablation in Liquid and Their Photocatalytic Activity. Nanotechnology 2009, 20, 1–7. [Google Scholar] [CrossRef]
- Siuzdak, K.; Sawczak, M.; Klein, M.; Nowaczyk, G.; Jurga, S.; Cenian, A. Preparation of Platinum Modified Titanium Dioxide Nanoparticles with the Use of Laser Ablation in Water. Phys. Chem. Chem. Phys. 2014, 16, 15199–15206. [Google Scholar] [CrossRef]
- Slavinskaya, E.M.; Stadnichenko, A.I.; Muravyov, V.V.; Kardash, T.Y.; Derevyannikova, E.A.; Zaikovskii, V.I.; Stomkus, O.A.; Lapin, I.N.; Svetlichnyi, V.A.; Boronin, A.I. Transformation of a Pt–CeO2 Mechanical Mixture of Pulsed-Laser-Ablated Nanoparticles to a Highly Active Catalyst for Carbon Monoxide Oxidation. ChemCatChem 2018, 10, 2232–2247. [Google Scholar] [CrossRef]
- Fedorova, E.A.; Stadnichenko, A.I.; Slavinskaya, E.M.; Kibis, L.S.; Stonkus, O.A.; Svintsitskiy, D.A.; Lapin, I.N.; Romanenko, A.V.; Svetlichnyi, V.A.; Boronin, A.I. A Study of PtAl2O3 Nanocomposites Obtained by Pulsed Laser Ablation to Be Used as Catalysts of Oxidation Reactions. J. Struct. Chem. 2020, 61, 332–346. [Google Scholar]
- Ogel, E.; Casapu, M.; Doronkin, D.E.; Popescu, R.; Störmer, H.; Mechler, C.; Marzun, G.; Barcikowski, S.; Türk, M.; Grunwaldt, J.-D. Impact of Preparation Method and Hydrothermal Aging on Particle Size Distribution of Pt/γ-Al2O3 and Its Performance in CO and NO Oxidation. J. Phys. Chem. C 2019, 123, 5433–5446. [Google Scholar] [CrossRef]
- TOPAS; Bruker AXS Inc.: Madison, WI, USA, 2009.
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data Analysis for X-Ray Absorption Spectroscopy Using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Bertoti, I.; Mohai, M.; Sullivan, J.L.; Saied, S.O. Surface Characterisation of Plasma-Nitrided Titanium: An XPS Study. Appl. Surf. Sci. 1995, 84, 357–371. [Google Scholar] [CrossRef]
- Svintsitskiy, D.A.; Kibis, L.S.; Stadnichenko, A.I.; Koscheev, S.V.; Zaikovskii, V.I.; Boronin, A.I. Highly Oxidized Platinum Nanoparticles Prepared through Radio-Frequency Sputtering: Thermal Stability and Reaction Probability towards CO. ChemPhysChem 2015, 16, 3318–3324. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. HISTORICAL Commentary NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Scarisoreanu, M.; Ilie, A.; Dutu, E.; Badoi, A.; Dumitrache, F.; Tanasa, E.; Mihailescu, C.N.; Mihailescu, I. Applied Surface Science Direct Nanocrystallite Size Investigation in Microstrained Mixed Phase TiO2 Nanoparticles by PCA of Raman Spectra. Appl. Surf. Sci. 2019, 470, 507–519. [Google Scholar] [CrossRef]
- Vovk, E.I.; Kalinkin, A.V.; Smirnov, M.Y.; Klembovskii, I.O.; Bukhtiyarov, V.I. XPS Study of Stability and Reactivity of Oxidized Pt Nanoparticles Supported on TiO2. J. Phys. Chem. C 2017, 121, 17297–17304. [Google Scholar] [CrossRef]
- Bruix, A.; Rodriguez, J.A.; Ramirez, P.J.; Senanayake, S.D.; Evans, J.; Park, J.B.; Stacchiola, D.; Liu, P.; Hrbek, J.; Illas, F. A New Type of Strong Metal − Support Interaction and the Production of H2 through the Transformation of Water on Pt/CeO2(111) and Pt/CeOx/TiO2(110) Catalysts. J. Am. Chem. Soc. 2012, 134, 8968–8974. [Google Scholar] [CrossRef]
- Lykhach, Y.; Kozlov, S.M.; Skála, T.; Tovt, A.; Stetsovych, V.; Tsud, N.; Dvořák, F.; Johánek, V.; Neitzel, A.; Mysliveček, J.; et al. Counting Electrons on Supported Nanoparticles. Nat. Mater. 2016, 15, 284–288. [Google Scholar] [CrossRef]
- Svintsitskiy, D.A.; Kibis, L.S.; Stadnichenko, A.I.; Slavinskaya, E.M.; Zaguzin, A.S.; Romanenko, A.V.; Derevyannikova, E.A.; Stonkus, O.A.; Boronin, A.I. The Influence of the Metal-Support Interaction on the Catalytic Activity of Pt/Al2O3 and Pt/TiO2 in NH3 Oxidation. AIP Conf. Proc. 2143 2019, 20028, 1–5. [Google Scholar] [CrossRef]
- Ivanova, E.; Mihaylov, M.; Daturi, M.; Hadjiivanov, K. FTIR Spectroscopy Study of CO and NO Adsorption and Co-Adsorption on Pt/TiO2. J. Mol. Catal. A Chem. 2007, 274, 179–184. [Google Scholar] [CrossRef]
- Seriani, N.; Pompe, W.; Ciacchi, L.C. Catalytic Oxidation Activity of Pt3O4 Surfaces and Thin Films. J. Phys. Chem. B 2006, 110, 14860–14869. [Google Scholar] [CrossRef] [PubMed]
- Fung, S.C. XPS Studies of Strong Metal-Support Interactions (SMSI)-Pt/TiO2. J. Catal. 1982, 76, 225–230. [Google Scholar] [CrossRef]
- Ioannides, T.; Verykios, X.E. Charge Transfer in Metal Catalysts Supported on Doped TiO2: A Theoretical Approach Based on Metal—Semiconductor Contact Theory. J. Catal. 1996, 161, 560–569. [Google Scholar] [CrossRef]
- Bahl, M.K.; Tsai, S.C.; Chung, Y.W. Auger and Photoemission Investigations of the Platinum-SrTiO3(100) Interface: Relaxation and Chemical-Shift Effects. Phys. Rev. B 1980, 21, 1344–1348. [Google Scholar] [CrossRef]
- Sexton, B.A.; Hughes, A.E.; Foger, K. XPS Investigation of Strong Metal-Support Interactions on Group IIIa-Va Oxides. J. Catal. 1982, 77, 85–93. [Google Scholar] [CrossRef]
- Moulder, J.F.; Strickle, W.F.; Sobol, P.E.; Bomben, K.D. A Reference Book of Standart Spectra for Identification and Interpretation of XPS Data; Chastain, J., Ed.; Perkin-Elmer Corporation Physical Electronics Division: Eden Prairie, MN, USA, 1992. [Google Scholar]
- Jehn, H. High Temperature Behaviour of Platinum Group Metals in Oxidizing Atmospheres. J. Less-Common Met. 1984, 100, 321–339. [Google Scholar] [CrossRef]
- Kraehnert, R.; Baerns, M. Kinetics of Ammonia Oxidation over Pt Foil Studied in a Micro-Structured Quartz-Reactor. Chem. Eng. J. 2008, 137, 361–375. [Google Scholar] [CrossRef]













© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).