The Transformation by Catalysis of Prebiotic Chemical Systems to Useful Biochemicals: A Perspective Based on IR Spectroscopy of the Primary Chemicals: I. The Synthesis of Peptides by the Condensation of Amino Acids


Abstract
:1. Introduction
2. The Selective Energy Transfer (SET) Theory
2.1. Peptide Bond Formation:
2.2. COS and Resonance Conditions
3. Further Tests of SET Catalysis
3.1. Iron(II/III) Hexacyano Complexes
3.2. Cyanamide Test
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Duve, C. Vital Dust: Life as a Cosmic Imperative; Basic Books: New York, NY, USA, 1995. [Google Scholar]
- Oparin, A.I. Origin of Life; Morgolis, T.S., Ed.; Dover Publications, Inc.: New York, NY, USA, 1953. [Google Scholar]
- Bernal, J.D. The Physical Basis of Life; Routledge & Kegan Paul: London, UK, 1951. [Google Scholar]
- Danger, G.; Plasson, R.; Pascal, R. Pathways for the formation and evolution of peptides in prebiotic environments. Chem. Soc. Rev. 2012, 41, 5416. [Google Scholar] [CrossRef] [PubMed]
- Holm, N.G.; Andersson, E. Hydrothermal Simulation Experiments as a Tool for Studies of the Origin of Life on Earth and Other Terrestrial Planets: A Review. Astrobiology 2005, 5, 444–460. [Google Scholar] [CrossRef] [PubMed]
- NIST Chemistry WebBook. Available online: http://webbook.nist.gov/chemistry (accessed on 14 November 2014).
- Herzberg, G. Molecular Spectra and Molecular Structure, II. Infrared and Raman Spectra of Polyatomic Molecules; D van Nostrand Company, Inc.: Princeton, NY, USA, 1962. [Google Scholar]
- Herzberg, G. Spectra of Diatomic Molecules, 2nd ed.; Van Norstrand Co., Inc.: New York, NY, USA, 1950. [Google Scholar]
- Colín-García, M.; Heredia, A.; Cordero, G.; Camprubí, A.; Negrón-Mendoza, A.; Ortega-Gutiérrez, F.; Beraldi, H.; Ramos-Bernal, S. Hydrothermal vents and prebiotic chemistry: A review. Boletín Soc. Geol. Mex. 2016, 68, 599–620. [Google Scholar] [CrossRef]
- Dodd, M.S.; Papineau, D.; Grenne, T.; Slack, J.F.; Rittner, M.; Pirajno, F.; O’Neil, J.; Little, C.T.S. Evidence for early life in Earth’s oldest hydrothermal vent precipitates. Nature 2017, 543, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.L. A Production of Amino Acids Under Possible Primitive Earth Conditions. Science 1953, 117, 528–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, S.L.; Urey, H.C. Organic compound synthesis on the primitive earth. Science 1959, 130, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Larsson, R. A model of selective energy transfer at the active site of the catalyst. J. Mol. Catal. 1989, 55, 70–83. [Google Scholar] [CrossRef]
- Larsson, R. A SET Approach to the Interplay of Catalysts and Reactants. Catalysts 2018, 8, 97. [Google Scholar] [CrossRef] [Green Version]
- Larsson, R. Concluding remarks on the theory of selective energy transfer and exemplification on a zeolite kinetics study. Monatsh. Chem. 2013, 144, 21–28. [Google Scholar] [CrossRef]
- Kittel, C.; Knight, W.D.; Ruderman, M.A. Mechanics, Berkeley Physics Course; McGraw-Hill: New York, NY, USA, 1965; Volume 1, Chapter 7. [Google Scholar]
- Larsson, R. Propane Dehydrogenation Catalyzed by ZSM-5 Zeolites. A Mechanistic Study Based on the Selective Energy Transfer (SET) Theory. Molecules 2015, 20, 2529–2535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, R. An analysis of ammonia synthesis by the model of Selective Energy Transfer (SET). Ann. Math. Phys. 2019, 2, 038–050. [Google Scholar] [CrossRef] [Green Version]
- James, W. Pragmatism; Longmans, Green and Co.: New York, NY, USA, 1955. [Google Scholar]
- Berzelius, J. Årsberättelse om Framstegen i Physik och Chemie; Royal Swedish Academy of Sciences: Stockholm, Sweden, 1835. [Google Scholar]
- Ostwald, W. Nobel Lecture; The Royal Swedish Academy of Sciences: Stockholm, Sweden, 1909. [Google Scholar]
- Leman, L.; Orgel, L.; Ghadiri, M.R. Carbonyl Sulfide-Mediated Prebiotic Formation of Peptides. Science 2004, 306, 283–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds, 4th ed.; John Wiley & Sons: New York, NY, USA, 1986; p. 236. [Google Scholar]
- Wikipedia. Available online: https://en.wikipedia.org/wiki/Molecular_vibrationout-of-plane_vibrations/Case_study/partly (accessed on 23 April 2016).
- Huber, C.; Eisenreich, W.; Hecht, S.; Wächtershäuser, G. A Possible Primordial Peptide Cycle. Science 2003, 301, 938–940. [Google Scholar] [CrossRef] [PubMed]
- Huber, C. Peptides by Activation of Amino Acids with CO on (Ni,Fe)S Surfaces: Implications for the Origin of Life. Science 1998, 281, 670–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, E.T.; Zhou, M.; Burton, A.S.; Glavin, D.P.; Dworkin, J.P.; Krishnamurthy, R.; Fernández, F.M.; Bada, J.L. A Plausible Simultaneous Synthesis of Amino Acids and Simple Peptides on the Primordial Earth. Angew. Chem. 2014, 126, 8270–8274. [Google Scholar] [CrossRef]
- Davies, M.; Jones, W.J. The Infrared Spectrum and Structure of Cyanamide and Dimethyl cyanamide. Trans. Farad. Soc. 1958, 54, 1454–1463. [Google Scholar] [CrossRef]
- Rosado, M.T.; Duarte, M.L.T.; Fausto, R. Vibrational spectra of acid and alkaline glycine salts. Vib. Spectrosc. 1998, 16, 35–54. [Google Scholar] [CrossRef]

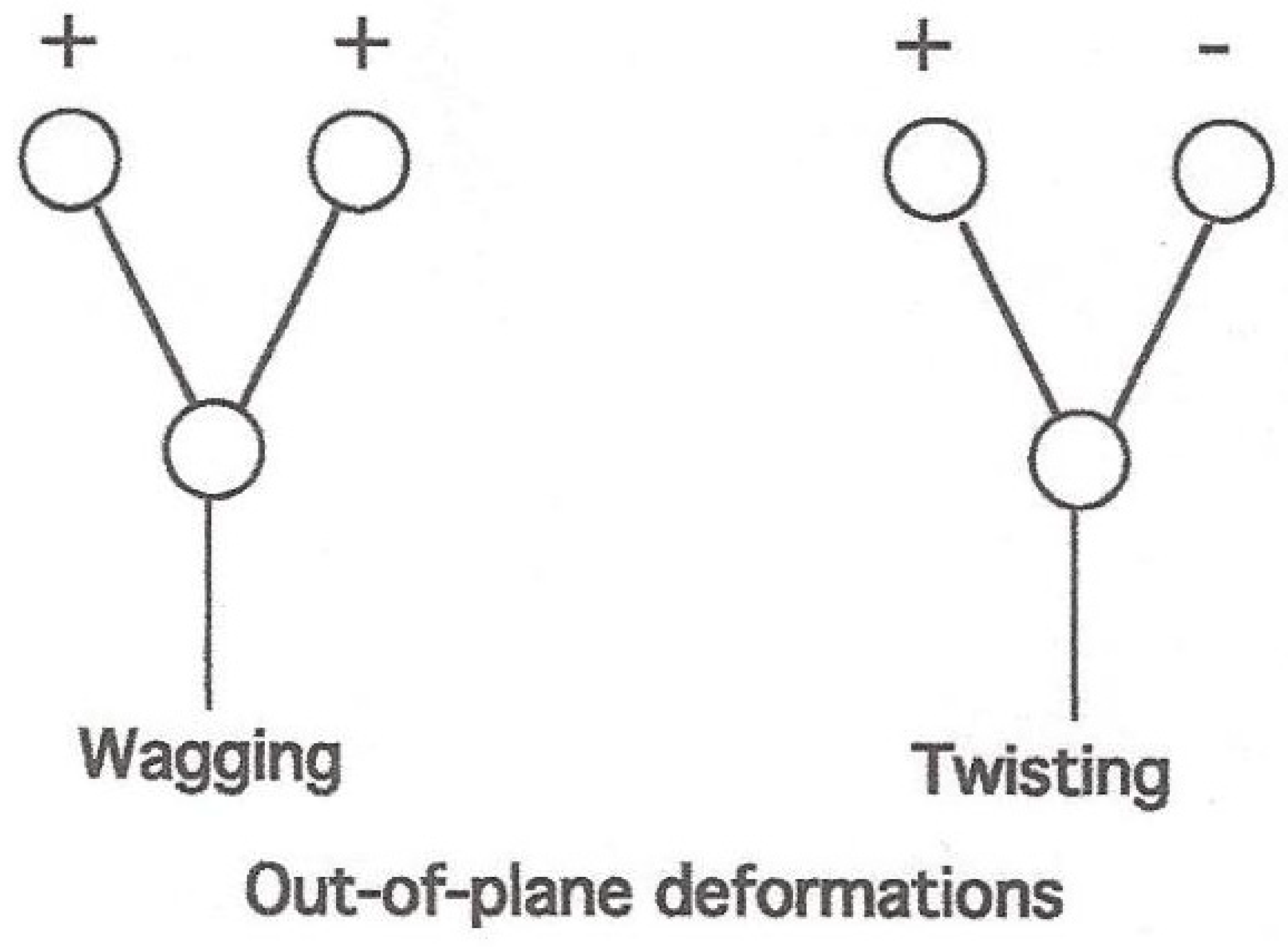





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assignment | Cu (Glycinate)2 | Ni (Glycinate)2 | Assignment | COS |
|---|---|---|---|---|
| υ (NH2) | 3320 | 3340 | ||
| υ (NH2) | 3260 | 3280 | υ 1 | 850 m |
| υ (C=O) | 1593 | 1589 | ||
| δ (NH2) | 1608 | 1610 | υ 2 | 527 m |
| υ (C–O) | 1392 | 1411 | ||
| ρt (NH2) | 1151 | 1095 | υ 3 | 2079 v s |
| ρw (NH2) | 1058 | 1038 | ||
| ρr (NH2) | 644 | 630 | ||
| δ (C=O) | 736 | 737 | ||
| ∏ (C=O) | 592 | 596 | ||
| υ (MN) | 439 | 439 | ||
| υ (MO) | 360 | 290 |
| Number | Cu (Glycinate)2 | Ways of Forming Differences | COS | Diff (Cu - COS) cm−1 | Diff Absolute Numbers % |
|---|---|---|---|---|---|
| 1 | 1593 | υ C - υ1(COS) | 850 | 743 | 46.6 |
| 2 | υCu - 2 × υ1(COS) | −107 | 6.7 | ||
| 3 | 1608 | υCu - υ1(COS) | 850 | 758 | 47.1 |
| 4 | υCu - 2 × υ1(COS) | −92 | 5.7 | ||
| 5 | 1392 | υCu - υ1(COS) | 850 | 542 | 38.9 |
| 6 | υCu - 2 × υ1(COS) | −308 | 22.1 | ||
| 7 | υCu - 2 × υ2(COS) | 527 | −338 | 24.3 | |
| 8 | υCu - 3 × υ2 (COS) | 1581 | −189 | 13.6 | |
| 9 | 1058 | υCu - 2 × υ2 (COS) | 2 × 527 | 4 | 0.4 |
| 10 | 1058 | 2 × υCu - υ 3(COS) | 2079 | 37 | 3.5 |
| Conditions | Glycine Vibration cm−1 [29] | Factor | Resulting Product cm−1 | Cyanamide Vibration cm−1 [28] | Diff% |
|---|---|---|---|---|---|
| Acid | Not present | ||||
| Neutral | 882 (m) | 1 | 882 | 937 (v w) | 6.2 |
| 2 | 1764 | 1576 (m) | 10.7 | ||
| 3 | 2646 | 2731 (v w) | 3.2 | ||
| Alkaline | 1069 (w) | 1 | 1069 | 1094 (w) | 2.3 |
| 2 | 2138 | 2199 (v w) | 2.9 | ||
| 3 | 3207 | 3261 (v s) | 1.7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larsson, R.; Malek, A. The Transformation by Catalysis of Prebiotic Chemical Systems to Useful Biochemicals: A Perspective Based on IR Spectroscopy of the Primary Chemicals: I. The Synthesis of Peptides by the Condensation of Amino Acids. Appl. Sci. 2020, 10, 928. https://doi.org/10.3390/app10030928
Larsson R, Malek A. The Transformation by Catalysis of Prebiotic Chemical Systems to Useful Biochemicals: A Perspective Based on IR Spectroscopy of the Primary Chemicals: I. The Synthesis of Peptides by the Condensation of Amino Acids. Applied Sciences. 2020; 10(3):928. https://doi.org/10.3390/app10030928
Chicago/Turabian StyleLarsson, Ragnar, and Abdul Malek. 2020. "The Transformation by Catalysis of Prebiotic Chemical Systems to Useful Biochemicals: A Perspective Based on IR Spectroscopy of the Primary Chemicals: I. The Synthesis of Peptides by the Condensation of Amino Acids" Applied Sciences 10, no. 3: 928. https://doi.org/10.3390/app10030928