Preparation of Microfibrillated Cellulose from Wood Pulp through Carbamate Modification and Colloid Milling


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of CC by a Solid Phase Method
2.3. Determination of Nitrogen Content—Kjeldahl Method
2.4. Preparation of CCNF by Colloid Milling
2.5. Zeta Potential Determination
2.6. Film-Forming of Starch/CCNF Nanocomposites
2.7. Characterization
2.7.1. Fourier Transform Infrared (FTIR) Spectroscopy
2.7.2. Nuclear Magnetic Resonance (NMR) Spectroscopy
2.7.3. X-ray Diffraction (XRD)
2.7.4. Scanning Electron Microscope (SEM)
2.7.5. Thermogravimetric Analysis (TGA)
2.7.6. Zeta Potential
2.7.7. Tensile Measurement
2.7.8. Statistical Analysis
3. Results and Discussion
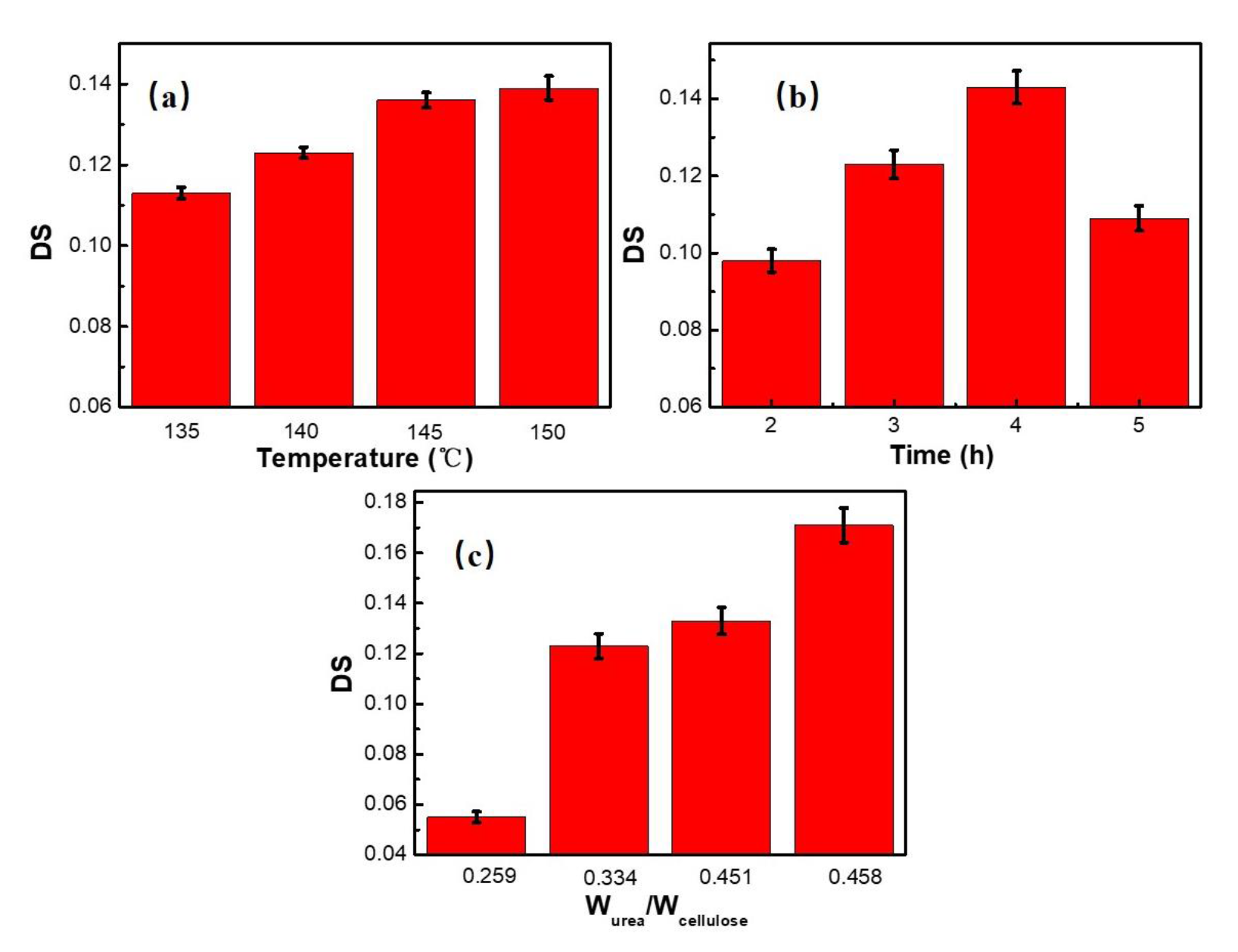
3.1. Preparation Conditions of CC
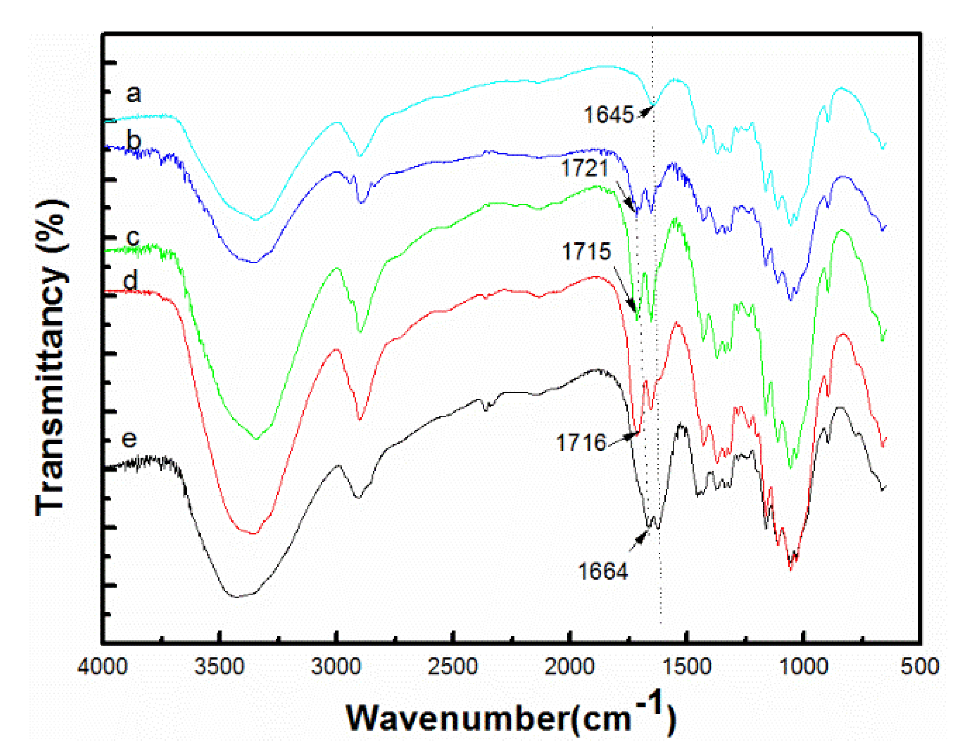
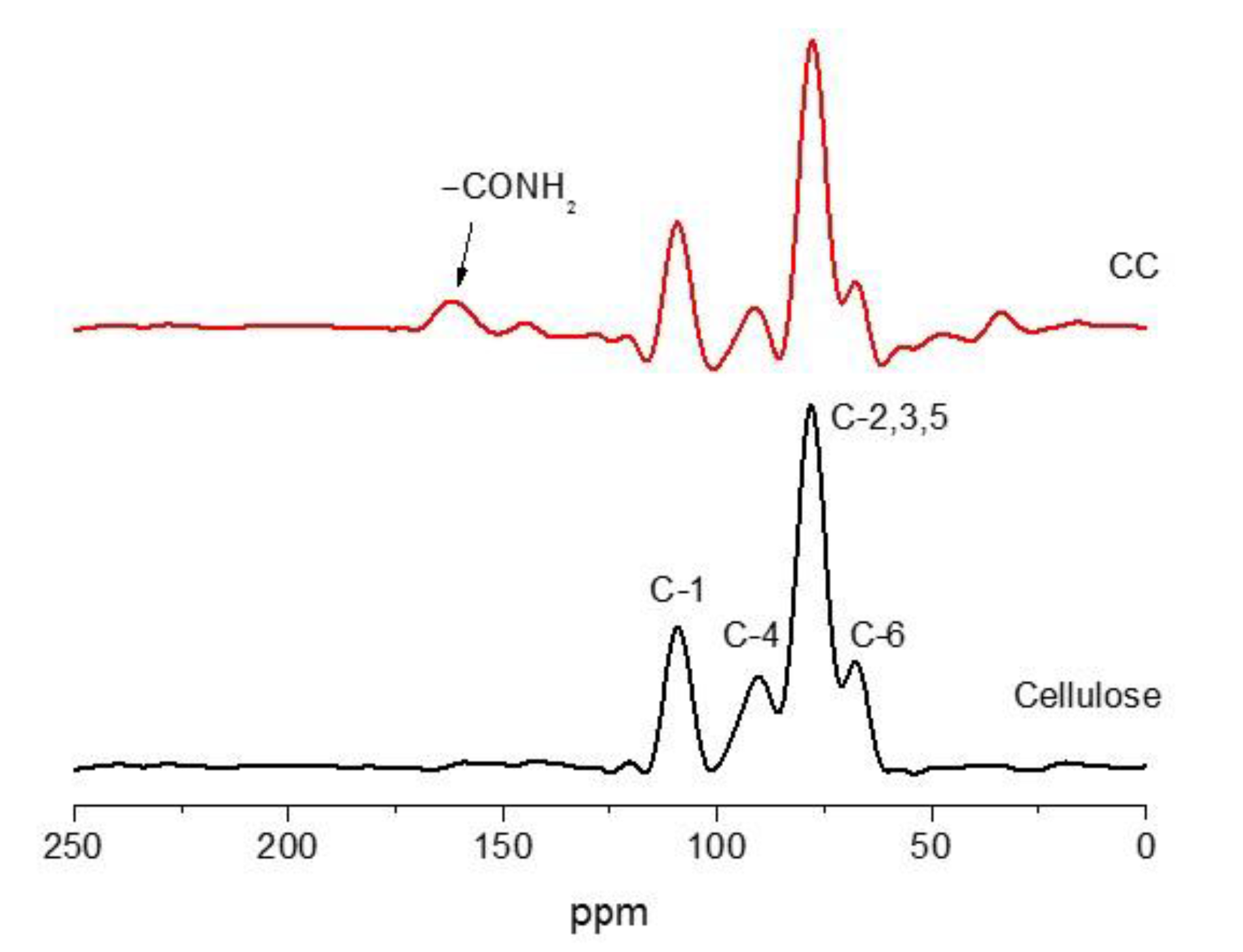
3.2. Confirmation of Carbamation of Cellulose
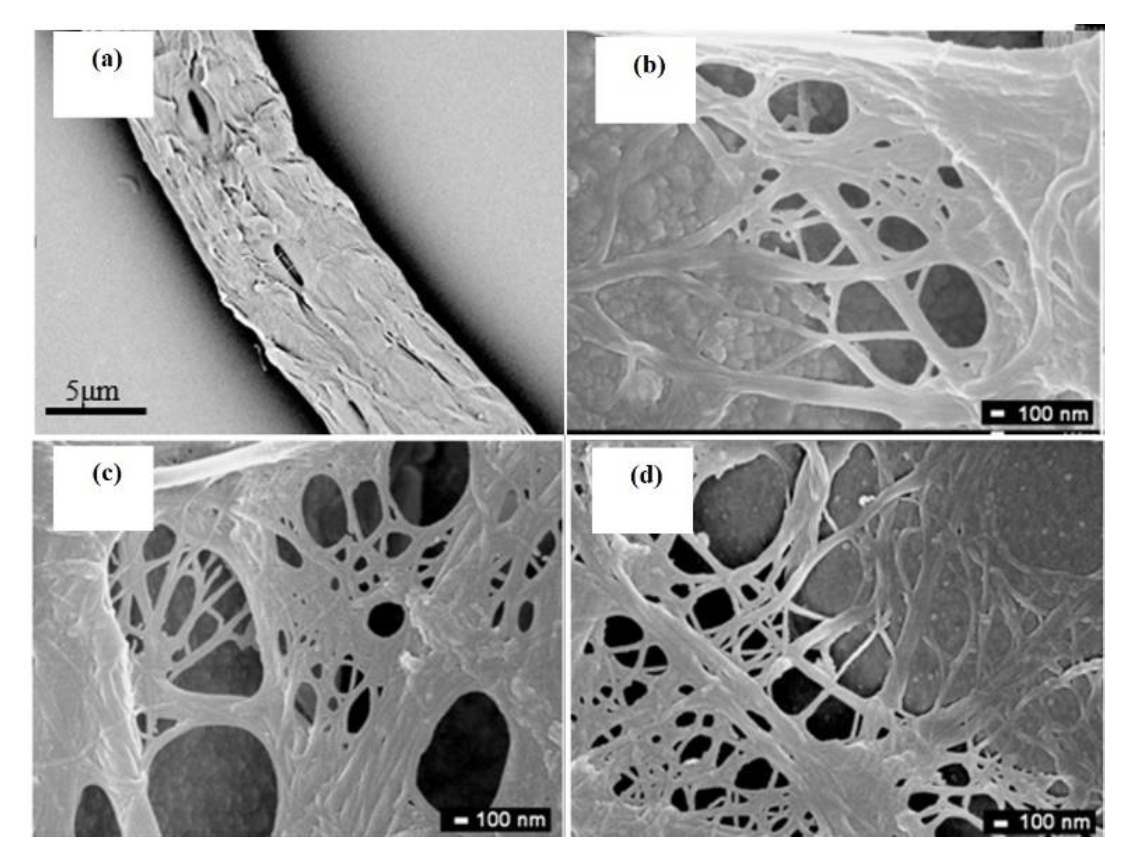
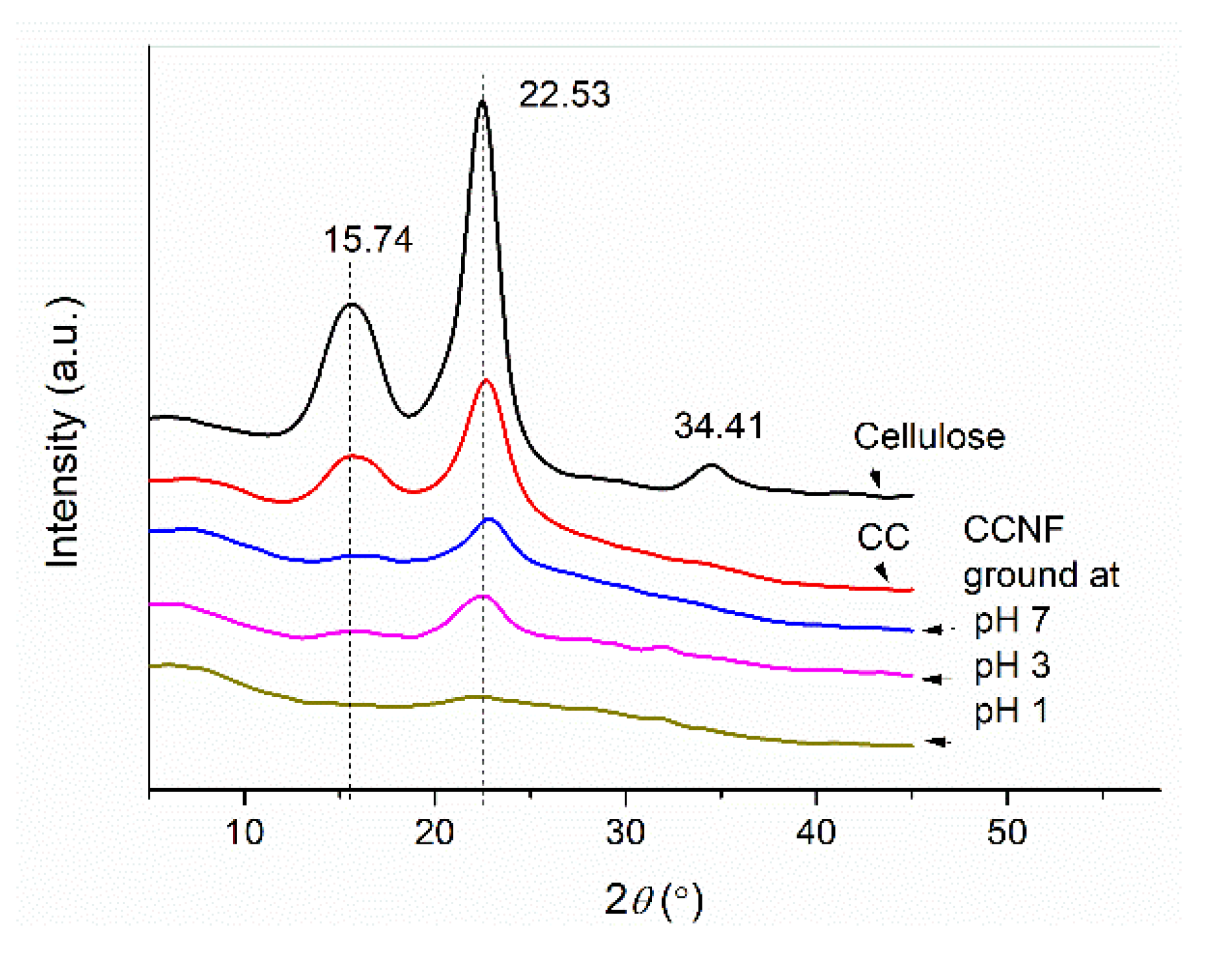
3.3. Preparation and Structures of CCNF
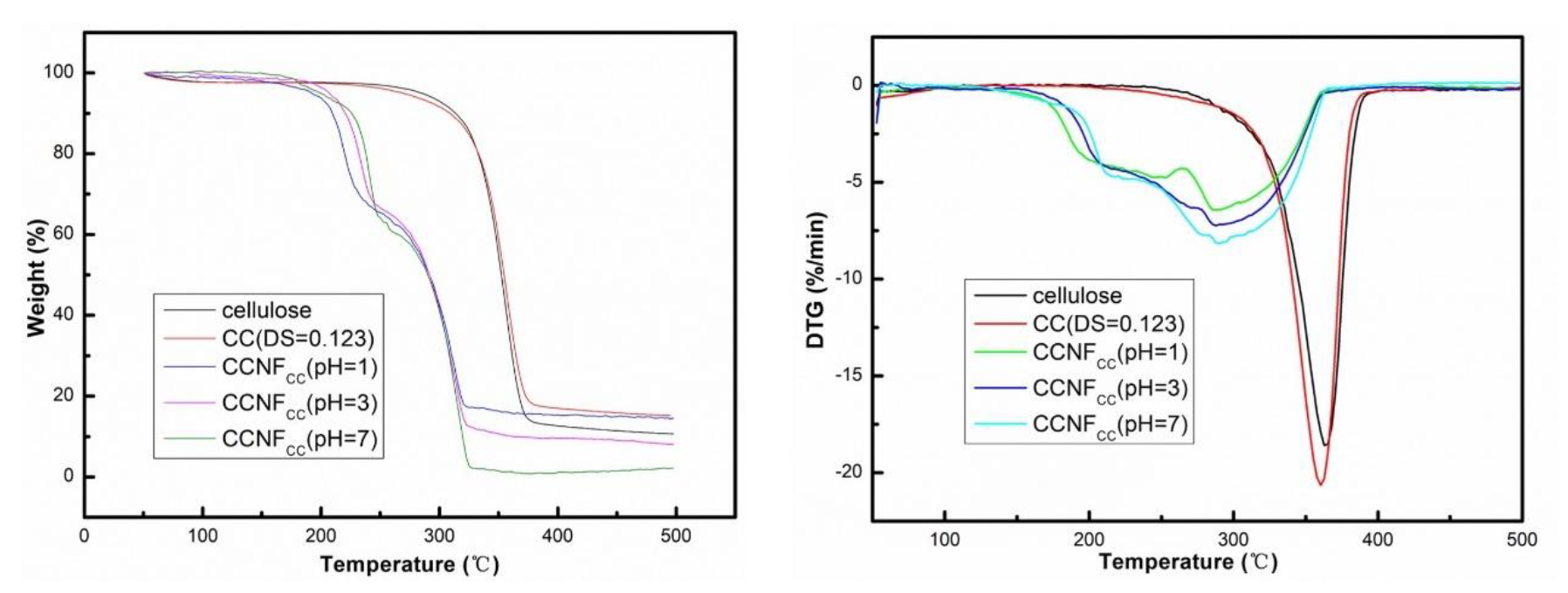
3.4. Thermal Properties of Cellulose and CC
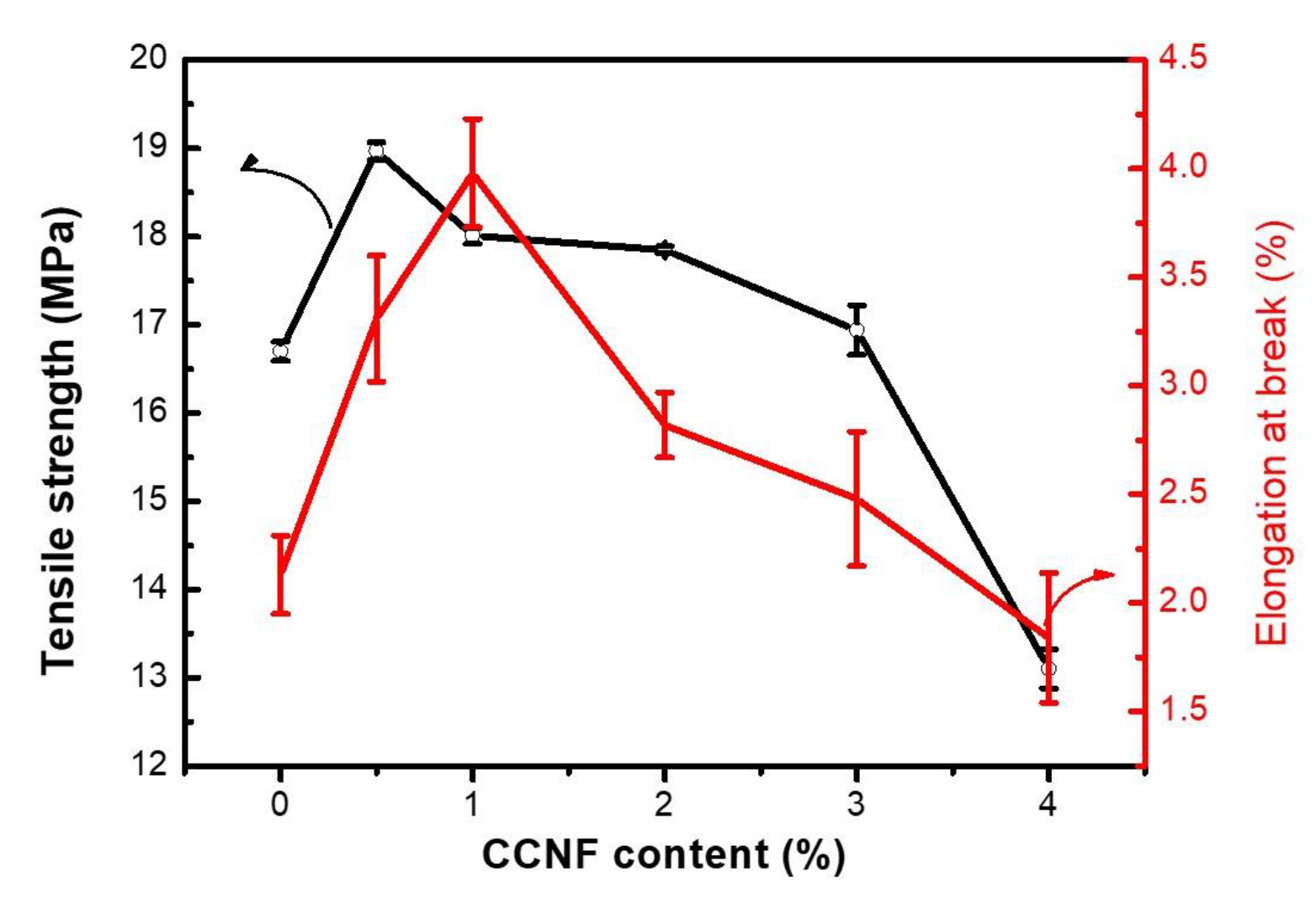
3.5. Mechanical Properties of CCNF/CS Nanocomposite Films
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gelfand, I.; Sahajpal, R.; Zhang, X.Z.; Izaurralde, R.C.; Gross, K.L.; Robertson, G.P. Sustainable bioenergy production from marginal lands in the US Midwest. Nature 2013, 493, 514–517. [Google Scholar] [CrossRef]
- Tian, Y.; Zhang, K.; Zhou, M.; Wei, Y.J.; Cheng, F.; Lin, Y.; Zhu, P.X. High-performance starch films reinforced with microcrystalline cellulose made from eucalyptus pulp via ball milling and mercerization. Starch/Stӓrke 2019, 71, 1800218. [Google Scholar] [CrossRef]
- Cheng, G.; Zhu, P.X.; Li, J.L.; Cheng, F.; Lin, Y.; Zhou, M. All-cellulose films with excellent strength and toughness via a facile approach of dissolution-regeneration. J. Appl. Polym. Sci. 2019, 136, 46925. [Google Scholar] [CrossRef]
- Borrega, M.; Orelma, H. Cellulose Nanofibril (CNF) Films and Xylan from Hot Water Extracted Birch Kraft Pulps. Appl. Sci. 2019, 9, 3436. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zheng, S.; Hu, Z.; Zhong, L.; Wang, Y.; Zhang, X.; Xue, J. Preparation of Polyvinyl Alcohol/Bacterial-Cellulose-Coated Biochar–Nanosilver Antibacterial Composite Membranes. Appl. Sci. 2020, 10, 752. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.; Zhou, M.; Henriksson, M.; Henriksson, G.; Berglund, L.; Lindström, T. An environmentally friendly method for enzyme-assisted preparation of microfibrillated cellulose (MFC) nanofibers. Eur. Polym. 2007, 43, 3434–3441. [Google Scholar]
- Li, J.; Wei, X.; Wang, Q.; Chen, J.; Chang, G.; Kong, L.; Su, J.; Liu, Y. Homogeneous isolation of nanocellulose from sugarcane bagasse by high pressure homogenization. Carbohydr. Polym. 2012, 90, 1609–1613. [Google Scholar] [CrossRef]
- Wang, Q.Q.; Zhu, J.Y.; Gleisner, R.; Kuster, T.A.; Baxa, U.; McNeil, S.E. Morphological development of cellulose fibrils of a bleached eucalyptus pulp by mechanical fibrillation. Cellulose 2012, 19, 1631–1643. [Google Scholar] [CrossRef]
- Khalil, H.A.; Bhat, A.H.; Yusra, A.I. Green composites from sustainable cellulose nanofibrils: A review. Carbohydr. Polym. 2012, 87, 963–979. [Google Scholar] [CrossRef]
- Siró, I.; Plackett, D. Microfibrillated cellulose and new nanocomposite materials: A review. Cellulose 2010, 17, 459–494. [Google Scholar] [CrossRef]
- Klemm, D.; Kramer, F.; Moritz, S.; Lindström, T.; Ankerfors, M.; Gray, D.; Dorris, A. Nanocelluloses: A new family of nature-based materials. Angew. Chem. Int. Ed. 2011, 50, 5438–5466. [Google Scholar] [CrossRef]
- Zimmermann, T.; Bordeanu, N.; Strub, E. Properties of nanofibrillated cellulose from different raw materials and its reinforcement potential. Carbohydr. Polym. 2010, 79, 1086–1093. [Google Scholar] [CrossRef]
- Dufresne, A.; Cavaille, J.Y.; Vignon, M.R. Mechanical behavior of sheets prepared from sugar beet cellulose microfibril. J. Appl. Polym. Sci. 1997, 64, 1185–1194. [Google Scholar] [CrossRef]
- Iwamoto, S.; Nakagaito, A.N.; Yano, H. Nano-fibrillation of pulp fibers for the processing of transparent nanocomposites. Appl. Phys. A Mater. Sci. Process. 2007, 89, 461–466. [Google Scholar] [CrossRef]
- Chakraborty, A.; Sain, M.; Kortschot, M. Cellulose microfibrils: A novel method of preparation using high shear refining and cryocrushing. Holzforschung 2005, 98, 102–107. [Google Scholar] [CrossRef]
- Alemdar, A.; Sain, M. Cellulose microfibrils: A novel method of preparation using high shear refining and cryocrushing. BioResour. Technol. 2008, 99, 1664–1671. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.K.; Zink-Sharp, A.; Renneckar, S.H.; Glasser, W.G. A new bio-based nanocomposite: Fibrillated TEMPO-oxidized celluloses in hydroxypropylcellulose matrix. Cellulose 2009, 16, 227–238. [Google Scholar] [CrossRef]
- Wang, S.; Cheng, Q. A novel process to isolate fibrils from cellulose fibers by high-intensity ultrasonication, part 1: Process optimization. J. Appl. Polym. Sci. 2009, 113, 1270–1275. [Google Scholar] [CrossRef]
- Lindström, T.; Aulin, C. Market and technical challenges and opportunities in the area of innovative new materials and composites based on nanocellulosics. Scand. J. For. Res. 2014, 29, 345–351. [Google Scholar] [CrossRef]
- Fukuzumi, H.; Saito, T.; Iwata, T.; Kumamoto, Y.; Isogai, A. Transparent and high gas barrier films of cellulose nanofibers prepared by TEMPO-mediated oxidation. Biomacromolecules 2009, 10, 162–165. [Google Scholar] [CrossRef]
- Klemm, D.; Heublein, B.; Fink, H.P.; Bohn, A. Cellulose: Fascinating biopolymer and sustainable raw material. Angew. Chem. Int. Ed. 2005, 44, 3358–3393. [Google Scholar] [CrossRef] [PubMed]
- Fu, F.; Zhou, J.; Zhou, X.; Zhang, L.N.; Li, D.; Kondo, T. Green Method for Production of Cellulose Multifilament from Cellulose Carbamate on a Pilot Scale. ACS Sustain. Chem. Eng. 2014, 2, 2363–2370. [Google Scholar] [CrossRef]
- Guo, Y.; Zhou, J.; Wang, Y.; Zhang, L.N.; Lin, X. An efficient transformation of cellulose into cellulose carbamates assisted by microwave irradiation. Cellulose 2010, 17, 1115–1125. [Google Scholar] [CrossRef]
- Gan, S.; Zakaria, S.; Chia, C.H.; Chen, R.S.; Ellis, A.V.; Kaco, H. Highly porous regenerated cellulose hydrogel and aerogel prepared from hydrothermal synthesized cellulose carbamate. PLoS ONE 2017, 12, e0173743. [Google Scholar] [CrossRef]
- Fu, F.; Xu, M.; Wang, H.; Wang, Y.; Ge, H.; Zhou, J. Improved synthesis of cellulose carbamates with minimum urea based on an easy scale-up method. ACS Sustain. Chem. Eng. 2015, 3, 1510–1517. [Google Scholar] [CrossRef]
- Zhang, Y.; Yin, C.; Zhang, Y.; Wu, H. Synthesis and characterization of cellulose carbamate from wood pulp, assisted by supercritical carbon dioxide. Bioresource 2013, 8, 1398–1408. [Google Scholar] [CrossRef]
- Paajanen, A.; Vaari, J. High-temperature decomposition of the cellulose molecule: A stochastic molecular dynamics study. Cellulose 2017, 24, 2713–2725. [Google Scholar] [CrossRef] [Green Version]
- French, A.D.; Cintrón, M.S. Cellulose polymorphy, crystallite size, and the Segal crystallinity index. Cellulose 2013, 20, 583–588. [Google Scholar] [CrossRef]
- Wang, D.; Hui, S.; Liu, C. Mass loss and evolved gas analysis in thermal decomposition of solid urea. Fuel 2017, 207, 268–273. [Google Scholar] [CrossRef]
- Brack, W.; Heine, B.; Birkhold, F.; Kruse, M.; Schoch, G.; Tischer, S.; Deutschmann, O. Kinetic modeling of urea decomposition based on systematic thermogravimetric analyses of urea and its most important by-products. Chem. Eng. Sci. 2014, 106, 1–8. [Google Scholar] [CrossRef]
- Zhou, H.; Long, Y.; Meng, A.; Chen, S.; Li, Q.; Zhang, Y. A novel method for kinetics analysis of pyrolysis of hemicellulose, cellulose, and lignin in TGA and macro-TGA. RSC Adv. 2015, 5, 26509–26516. [Google Scholar] [CrossRef]
- Labafzadeh, S.R.; Kavakka, J.S.; Vyavaharkar, K.; Sievänen, K.; Kilpeläinen, I. Preparation of cellulose and pulp carbamates through a reactive dissolution approach. RSC Adv. 2014, 4, 22434–22441. [Google Scholar] [CrossRef]
- Fu, F.; Yang, Q.; Zhou, J.; Hu, H.; Jia, B.; Zhang, L.N. Structure and properties of regenerated cellulose filaments prepared from cellulose carbamate–NaOH/ZnO aqueous solution. ACS Sustain. Chem. Eng. 2014, 2, 2604–2612. [Google Scholar] [CrossRef]
- Guo, Y.; Zhou, J.; Song, Y.; Zhang, L.N. An efficient and environmentally friendly method for the synthesis of cellulose carbamate by microwave heating. Macromol. Rapid Commun. 2009, 30, 1504–1508. [Google Scholar] [CrossRef]
- Nechyporchuk, O.; Pignon, F.; Belgacem, M.N. Morphological properties of nanofibrillated cellulose produced using wet grinding as an ultimate fibrillation process. J. Mater. Sci. 2015, 50, 531–541. [Google Scholar] [CrossRef]
- Kurouchi, H.; Kawamoto, K.; Sugimoto, H.; Nakamura, S.; Otani, Y.; Ohwada, T. Activation of electrophilicity of stable Y-delocalized carbamate cations in intramolecular aromatic substitution reaction: Evidence for formation of diprotonated carbamates leading to generation of isocyanates. J. Org. Chem. 2012, 77, 9313–9328. [Google Scholar] [CrossRef]
- Olszewska, A.; Eronen, P.; Johansson, L.S.; Malho, J.M.; Ankerfors, M.; Lindström, T.; Ruokolainen, J.; Laine, J.; Österberg, M. The behaviour of cationic nanofibrillar cellulose in aqueous media. Cellulose 2011, 18, 1213–1226. [Google Scholar] [CrossRef]
- Oudiani, A.E.; Chaabouni, Y.; Msahli, S.; Sakli, F. Crystal transition from cellulose I to cellulose II in NaOH treated Agave americana L. fibre. Carbohydr. Polym. 2011, 86, 1221–1229. [Google Scholar] [CrossRef]
- Ishikawa, A.; Okano, T. Fine structure and tensile properties of ramie fibres in the crystalline form of cellulose I, II, IIII and IVI. Polymer 1997, 38, 463–468. [Google Scholar] [CrossRef]
- Oh, S.Y.; Yoo, D.I.; Shin, Y.; Kim, H.C.; Kim, H.Y.; Chung, Y.S.; Park, W.H.; Youk, J.H. Crystalline structure analysis of cellulose treated with sodium hydroxide and carbon dioxide by means of X-ray diffraction and FTIR spectroscopy. Carbohydr. Res. 2005, 340, 2376–2391. [Google Scholar] [CrossRef]
- Hubbe, M.A.; Rojas, O.J.; Lucia, L.A.; Mohini, S. Cellulosic nanocomposites: A review. BioResource 2008, 3, 929. [Google Scholar]
- Cheng, G.; Zhou, M.; Wei, Y.J.; Cheng, F.; Zhu, P.X. Comparison of mechanical reinforcement effects of cellulose nanocrystal, cellulose nanofiber and microfibrillated cellulose in starch composites. Polym. Compos. 2019, 40, E365–E372. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wurea/Wcell | Temperature (°C) | Time (h) | DS |
|---|---|---|---|
| 0.334 | 135 | 3 | 0.113 |
| 0.334 | 140 | 3 | 0.123 |
| 0.334 | 145 | 3 | 0.136 |
| 0.334 | 150 | 3 | 0.139 |
| 0.334 | 140 | 2 | 0.098 |
| 0.334 | 140 | 4 | 0.143 |
| 0.334 | 140 | 5 | 0.109 |
| 0.259 | 140 | 3 | 0.055 |
| 0.451 | 140 | 3 | 0.133 |
| 0.485 | 140 | 3 | 0.171 |
| Sample a | Zeta Potential (mV) | Crystallinity (%) | |
|---|---|---|---|
| Cellulose | - | 68.04 ± 3.40 | |
| CC | - | 30.81 ± 1.23 | |
| CC groundat pH | 7 | 3.90 ± 0.04 | 10.77 ± 0.22 |
| 3 | 8.97 ± 0.09 | 8.78 ± 0.26 | |
| 1 | 16.49 ± 0.10 | 2.27 ± 0.11 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, Y.; Zhou, M.; Yao, A.; Zhu, P. Preparation of Microfibrillated Cellulose from Wood Pulp through Carbamate Modification and Colloid Milling. Appl. Sci. 2020, 10, 1977. https://doi.org/10.3390/app10061977
Wei Y, Zhou M, Yao A, Zhu P. Preparation of Microfibrillated Cellulose from Wood Pulp through Carbamate Modification and Colloid Milling. Applied Sciences. 2020; 10(6):1977. https://doi.org/10.3390/app10061977
Chicago/Turabian StyleWei, Yujun, Mi Zhou, Anrong Yao, and Puxin Zhu. 2020. "Preparation of Microfibrillated Cellulose from Wood Pulp through Carbamate Modification and Colloid Milling" Applied Sciences 10, no. 6: 1977. https://doi.org/10.3390/app10061977