Hydrogen Inter-Cage Hopping and Cage Occupancies inside Hydrogen Hydrate: Molecular-Dynamics Analysis


Abstract
:1. Introduction
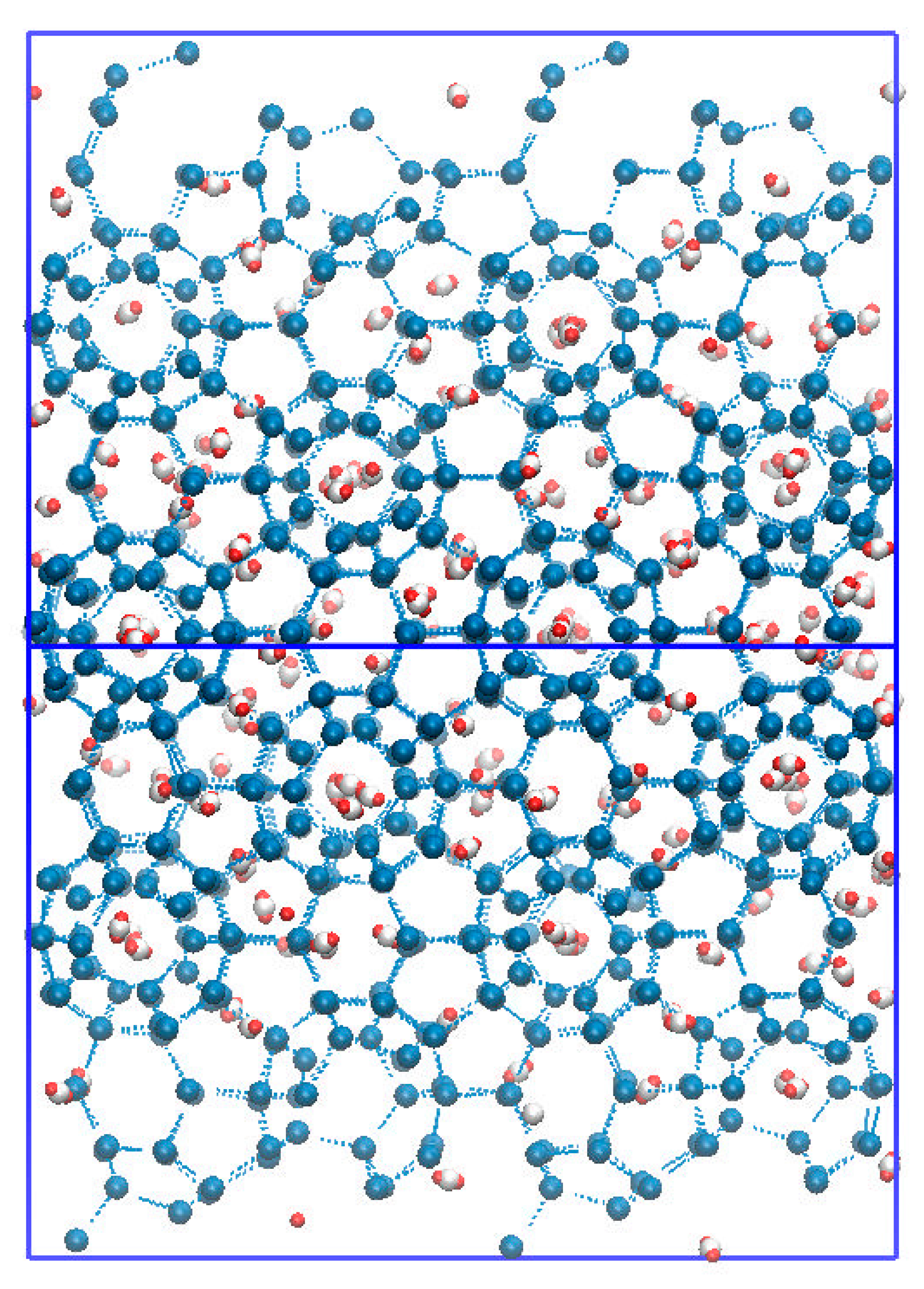
2. Computational Details
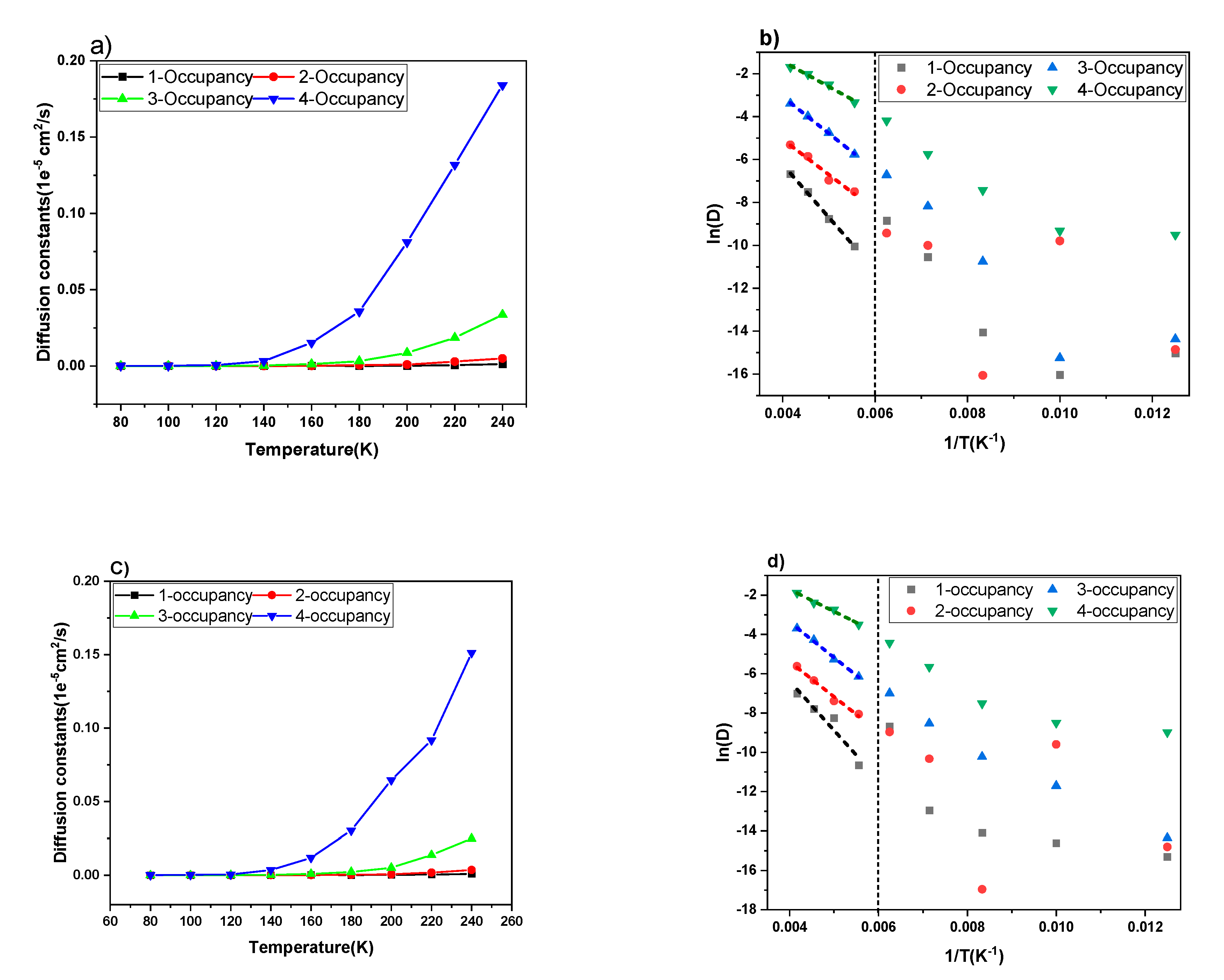
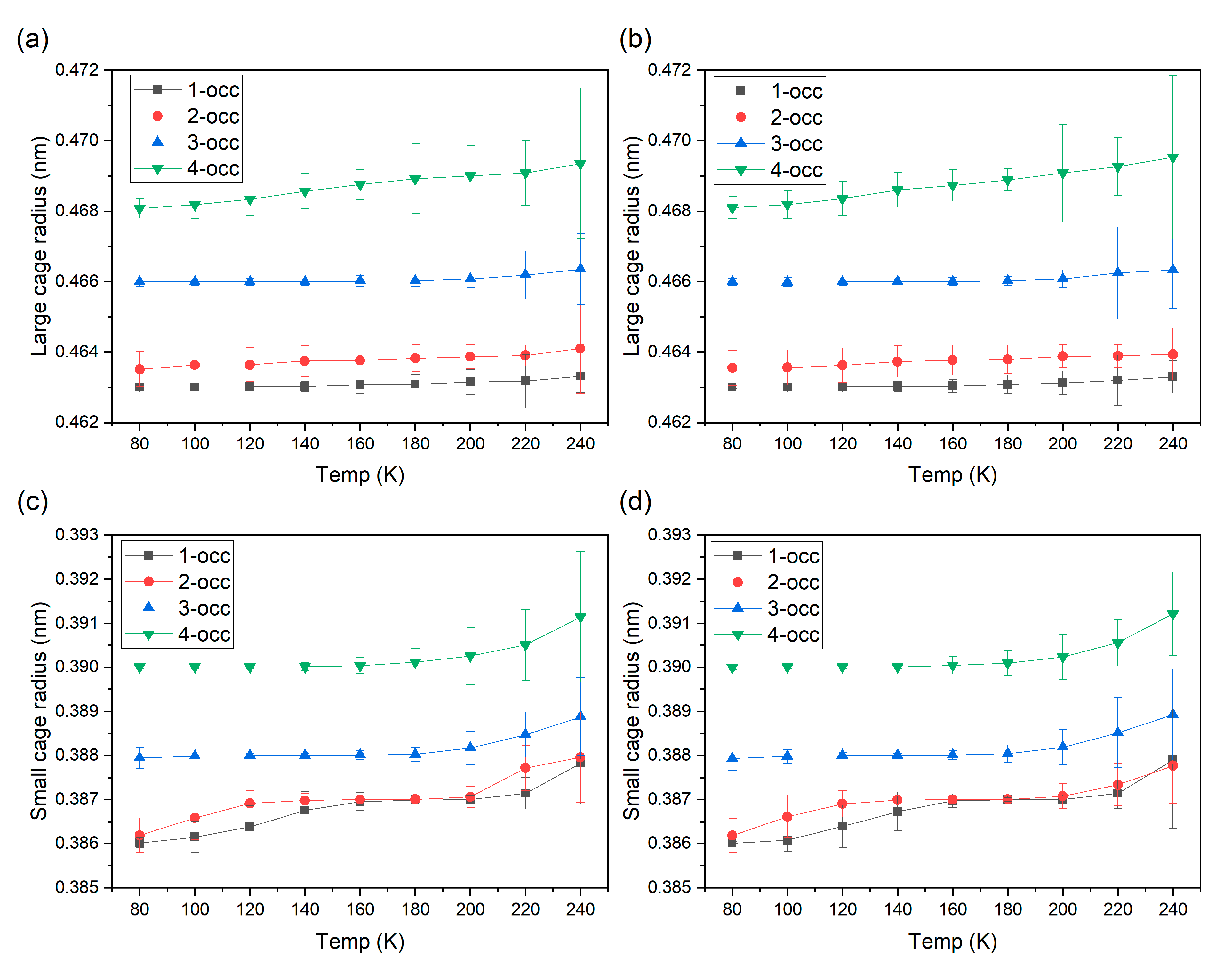
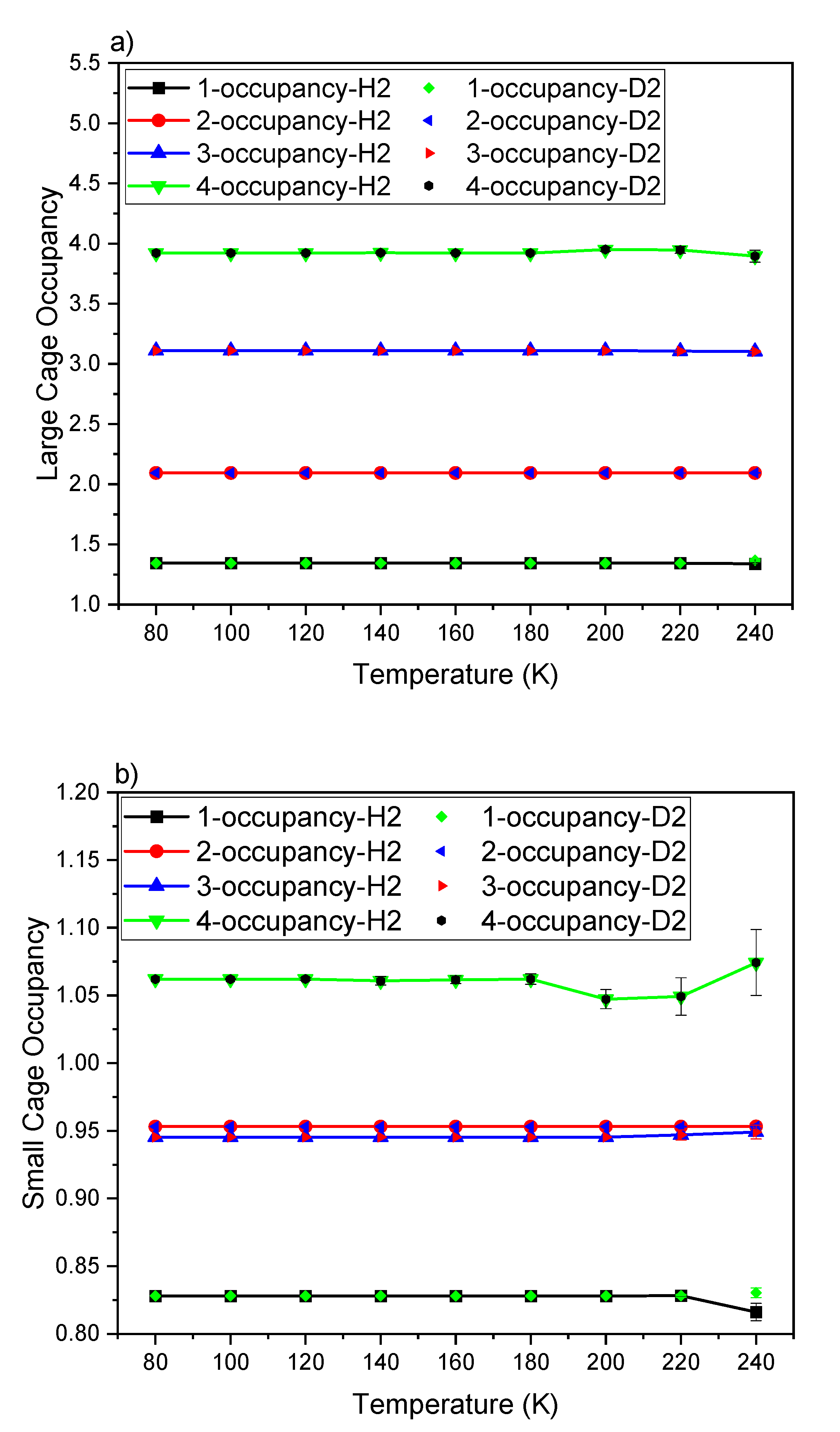
3. Result and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Al-Juboori, O.; Sher, F.; Khalid, U.; Niazi, M.B.K.; Chen, G.Z. Electrochemical Production of Sustainable Hydrocarbon Fuels from CO2 Co-electrolysis in Eutectic Molten Melts. ACS Sustain. Chem. Eng. 2020, 8, 12877–12890. [Google Scholar] [CrossRef]
- Al-Juboori, O.; Sher, F.; Hazafa, A.; Khan, M.K.; Chen, G.Z. The effect of variable operating parameters for hydrocarbon fuel formation from CO2 by molten salts electrolysis. J. CO2 Util. 2020, 40, 101193. [Google Scholar] [CrossRef]
- Al-Shara, N.K.; Sher, F.; Yaqoob, A.; Chen, G.Z. Electrochemical investigation of novel reference electrode Ni/Ni(OH)2 in comparison with silver and platinum inert quasi-reference electrodes for electrolysis in eutectic molten hydroxide. Int. J. Hydrogen Energy 2019, 44, 27224–27236. [Google Scholar] [CrossRef]
- Al-Shara, N.K.; Sher, F.; Iqbal, S.Z.; Curnick, O.; Chen, G.Z. Design and optimization of electrochemical cell potential for hydrogen gas production. J. Energy Chem. 2020, 52, 421–427. [Google Scholar] [CrossRef]
- Al-Shara, N.K.; Sher, F.; Iqbal, S.Z.; Sajid, Z.; Chen, G.Z. Electrochemical study of different membrane materials for the fabrication of stable, reproducible and reusable reference electrode. J. Energy Chem. 2020, 49, 33–41. [Google Scholar] [CrossRef]
- Sher, F.; Al-Shara, N.K.; Iqbal, S.Z.; Jahan, Z.; Chen, G.Z. Enhancing hydrogen production from steam electrolysis in molten hydroxides via selection of non-precious metal electrodes. Int. J. Hydrogen Energy 2020, 45, 28260–28271. [Google Scholar] [CrossRef]
- Züttel, A. Hydrogen storage methods. Naturwissenschaften 2004, 91, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Fichtner, M. Nanotechnological aspects in materials for hydrogen storage. Adv. Eng. Mater. 2005, 7, 443–455. [Google Scholar] [CrossRef]
- Mao, W.L.; Mao, H.; Goncharov, A.F.; Struzhkin, V.V.; Guo, Q.; Hu, J.; Shu, J.; Hemley, R.J.; Somayazulu, M.; Zhao, Y. Hydrogen Clusters in Clathrate Hydrate. Science 2002, 297, 2247–2249. [Google Scholar] [CrossRef]
- Humphry Davy, I. The Bakerian Lecture. On some of the combinations of oxymuriatic gas and oxygene, and on the chemical relations of these principles, to inflammable bodies. Philos. Trans. R. Soc. Lond. 1811, 101, 1–35. [Google Scholar] [CrossRef]
- Krishnan, Y.; Ghaani, M.R.; English, N.J. Electric-Field Control of Neon Uptake and Release to and from Clathrate Hydrates. J. Phys. Chem. C 2019. [Google Scholar] [CrossRef]
- Alavi, S.; Ripmeester, J.A. Hydrogen-gas migration through clathrate hydrate cages. Angew. Chem. Int. Ed. 2007, 46, 6102–6105. [Google Scholar] [CrossRef] [Green Version]
- Ma, R.; Zhong, H.; Liu, J.; Zhong, J.; Yan, Y.; Zhang, J.; Xu, J. Molecular insights into Cage Occupancy of hydrogen hydrate: A computational study. Processes 2019, 7, 699. [Google Scholar] [CrossRef] [Green Version]
- Ghaani, M.R.; English, N.J. Non-equilibrium molecular-dynamics study of electromagnetic-field-induced propane-hydrate dissociation. J. Chem. Phys. 2018, 149. [Google Scholar] [CrossRef]
- Ghaani, M.R.; Catti, M. Dehydrogenation properties of the LiNH2BH3/MgH2 and LiNH2BH3/LiBH4 bi-component hydride systems for hydrogen storage applications. Mater. Renew. Sustain. Energy 2018, 7, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, T.; Brumby, P.E.; Yasuoka, K.; Sum, A.K. Mechanism for H 2 diffusion in sII hydrates by molecular dynamics simulations. J. Chem. Phys. 2020, 153, 054706. [Google Scholar] [CrossRef]
- English, N.J.; MacElroy, J.M.D. Perspectives on molecular simulation of clathrate hydrates: Progress, prospects and challenges. Chem. Eng. Sci. 2015, 121, 133–156. [Google Scholar] [CrossRef]
- Erfan-Niya, H.; Modarress, H.; Zaminpayma, E. Computational study on the structure II clathrate hydrate of methane and large guest molecules. J. Incl. Phenom. Macrocycl. Chem. 2011, 70, 227–239. [Google Scholar] [CrossRef]
- Loveday, J.S.; Nelmes, R.J. High-pressure gas hydrates. Phys. Chem. Chem. Phys. 2008, 10, 937–950. [Google Scholar] [CrossRef]
- Sloan, E.D., Jr.; Koh, C.A. Clathrate Hydrates of Natural Gases; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Falenty, A.; Hansen, T.C.; Kuhs, W.F. Formation and properties of ice XVI obtained by emptying a type sII clathrate hydrate. Nature 2014, 516, 231–233. [Google Scholar] [CrossRef]
- Gies, H.; Liebau, F.; Gerke, H. “Dodecasile”—Eine neue Reihe polytyper Einschlußverbindungen von SiO2. Angew. Chem. 2006, 94, 214–215. [Google Scholar] [CrossRef]
- Guloy, A.M.; Ramlau, R.; Tang, Z.; Schnelle, W.; Baitinger, M.; Grin, Y. A guest-free germanium clathrate. Nature 2006, 443, 320–323. [Google Scholar] [CrossRef] [PubMed]
- Gryko, J.; McMillan, P.F.; Marzke, R.F.; Ramachandran, G.K.; Patton, D.; Deb, S.K.; Sankey, O.F. Low-density framework form of crystalline silicon with a wide optical band gap. Phys. Rev. B Condens. Matter Mater. Phys. 2000, 62, R7707–R7710. [Google Scholar] [CrossRef]
- Alavi, S.; Ripmeester, J.A.; Klug, D.D. Molecular-dynamics study of structure II hydrogen clathrates. J. Chem. Phys. 2005, 123. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Alavi, S.; Shin, K.; Ripmeester, J.A. Molecular dynamics simulations of hydrogen bonding in clathrate hydrates with ammonia and methanol guest molecules. J. Chem. Eng. Data 2015, 60, 389–397. [Google Scholar] [CrossRef] [Green Version]
- Nath Chakraborty, S.; Gelb, L.D. A Monte Carlo simulation study of methane clathrate hydrates confined in slit-shaped pores. J. Phys. Chem. B 2012, 116, 2183–2197. [Google Scholar] [CrossRef]
- Huang, Y.; Zhu, C.; Wang, L.; Cao, X.; Su, Y.; Jiang, X.; Meng, S.; Zhao, J.; Zeng, X.C. A new phase diagram of water under negative pressure: The rise of the lowest-density clathrate s-III. Sci. Adv. 2016, 2, e1501010. [Google Scholar] [CrossRef] [Green Version]
- Bekker, H.; Berendsen, H.J.C.; Van Drunen, R.; Spoel, D. Van Der Gromacs: A parallel computer for molecular dynamics simulations. Phys. Comput. 1993, 92, 252–256. [Google Scholar]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B.; van der Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Model. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghesquière, P.; Mineva, T.; Talbi, D.; Theulé, P.; Noble, J.A.; Chiavassa, T. Diffusion of molecules in the bulk of a low density amorphous ice from molecular dynamics simulations. Phys. Chem. Chem. Phys. 2015, 17, 11455–11468. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; English, N.J.; MacElroy, J.M.D. Diffusive hydrogen inter-cage migration in hydrogen and hydrogen-tetrahydrofuran clathrate hydrates. J. Chem. Phys. 2013, 138, 094507. [Google Scholar] [CrossRef]
- Burnham, C.J.; English, N.J. Free-energy calculations of the intercage hopping barriers of hydrogen molecules in clathrate hydrates. J. Phys. Chem. C 2016, 120, 16561–16567. [Google Scholar] [CrossRef]
- Lu, Q.; He, X.; Hu, W.; Chen, X.; Liu, J. Stability, Vibrations, and Diffusion of Hydrogen Gas in Clathrate Hydrates: Insights from Ab Initio Calculations on Condensed-Phase Crystalline Structures. J. Phys. Chem. C 2019, 123, 12052–12061. [Google Scholar] [CrossRef]
- Ferdows, M.; Ota, M. Molecular simulation study for CO2 clathrate hydrate. Chem. Eng. Technol. 2005, 28, 168–173. [Google Scholar] [CrossRef]
- Matsui, T.; Hirata, M.; Yagasaki, T.; Matsumoto, M.; Tanaka, H. Communication: Hypothetical ultralow-density ice polymorphs. J. Chem. Phys. 2017, 147. [Google Scholar] [CrossRef]
- Huang, Y.; Zhu, C.; Wang, L.; Zhao, J.; Zeng, X.C. Prediction of a new ice clathrate with record low density: A potential candidate as ice XIX in guest-free form. Chem. Phys. Lett. 2017, 671, 186–191. [Google Scholar] [CrossRef]
- Kosyakov, V.I.; Shestakov, V.A. On the possibility of the existence of a new ice phase under negative pressures. Dokl. Phys. Chem. 2001, 376, 49–51. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, Y.; Zhu, C.; Li, H.; Zhao, J.; Wang, L.; Ojamäe, L.; Francisco, J.S.; Zeng, X.C. An ultralow-density porous ice with the largest internal cavity identified in the water phase diagram. Proc. Natl. Acad. Sci. USA 2019, 116, 12684–12691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baia, J.; Angellb, C.A.; Zenga, X.C. Guest-free monolayer clathrate and its coexistence with two-dimensional high-density ice. Proc. Natl. Acad. Sci. USA 2010, 107, 5718–5722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conde, M.M.; Vega, C.; Tribello, G.A.; Slater, B. The phase diagram of water at negative pressures: Virtual ices. J. Chem. Phys. 2009, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desmedt, A.; Lechner, R.E.; Lassegues, J.-C.; Guillaume, F.; Cavagnat, D.; Grondin, J. Hydronium dynamics in the perchloric acid clathrate hydrate. Solid State Ionics 2013, 252, 19–25. [Google Scholar] [CrossRef]
- Desmedt, A.; Stallmach, F.; Lechner, R.E.; Cavagnat, D.; Lassègues, J.-C.; Guillaume, F.; Grondin, J.; Gonzalez, M.A. Proton dynamics in the perchloric acid clathrate hydrate HClO4⋅5.5H2O. J. Chem. Phys. 2004, 121, 11916–11926. [Google Scholar] [CrossRef] [PubMed]
- Desmedt, A.; Martin-Gondre, L.; Nguyen, T.T.; Pétuya, C.; Barandiaran, L.; Babot, O.; Toupance, T.; Grim, R.G.; Sum, A.K. Modifying the Flexibility of Water Cages by Co-Including Acidic Species within Clathrate Hydrate. J. Phys. Chem. C 2015, 119, 8904–8911. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Pétuya, C.; Talaga, D.; Desmedt, A. Promoting the Insertion of Molecular Hydrogen in Tetrahydrofuran Hydrate With the Help of Acidic Additives. Front. Chem. 2020, 8. [Google Scholar] [CrossRef]
- Brumby, P.E.; Yuhara, D.; Hasegawa, T.; Wu, D.T.; Sum, A.K.; Yasuoka, K. Cage occupancies, lattice constants, and guest chemical potentials for structure II hydrogen clathrate hydrate from Gibbs ensemble Monte Carlo simulations. J. Chem. Phys. 2019, 150, 134503. [Google Scholar] [CrossRef]

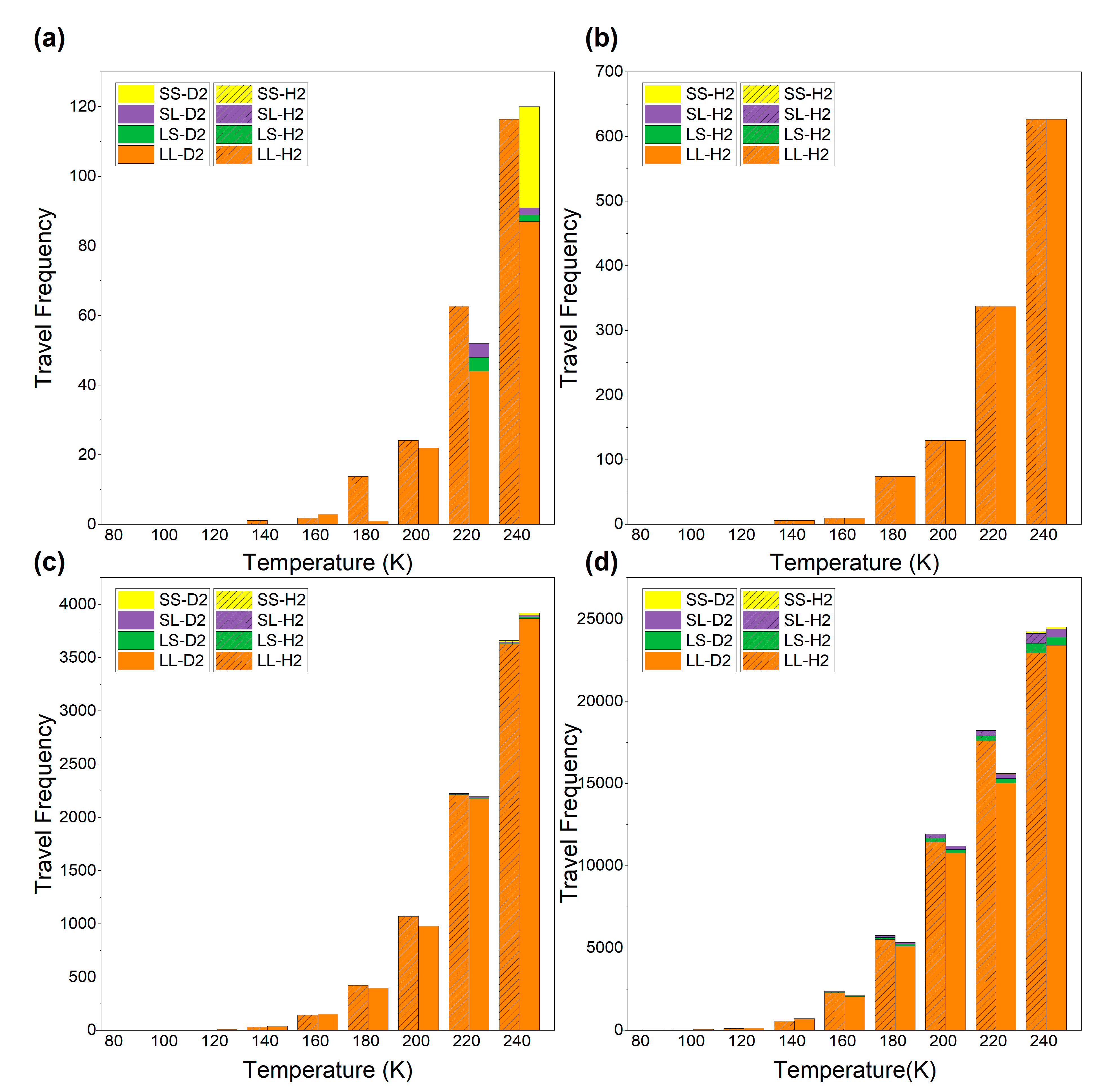

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
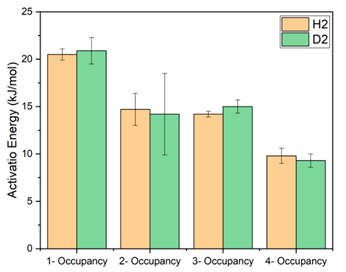
| Configuration | Activation Energy (kJ/mol) | |
|---|---|---|
| H2 | D2 | |
| 1-Occupancy | 20.5 ± 0.6 | 20.9 ± 1.4 |
| 2-Occupancy | 14.7 ± 1.7 | 14.2 ± 4.3 |
| 3-Occupancy | 14.2 ± 0.3 | 15.0 ± 0.7 |
| 4-Occupancy | 9.8 ± 0.8 | 9.3 ± 0.7 |
| Cage Type | Structure | Travel Possibility/Cage | Cage Number/Unit Cell | Travel Possibility/Unit Cell | ||||
|---|---|---|---|---|---|---|---|---|
| LL | SL/LS | SS | LL | SL/LS | SS | |||
| Large | 51264 | 4 | 12 | - | 8 | 14.28% | 42.85% | 42.85% |
| Small | 512 | - | 6 | 16 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krishnan, Y.; Ghaani, M.R.; Desmedt, A.; English, N.J. Hydrogen Inter-Cage Hopping and Cage Occupancies inside Hydrogen Hydrate: Molecular-Dynamics Analysis. Appl. Sci. 2021, 11, 282. https://doi.org/10.3390/app11010282
Krishnan Y, Ghaani MR, Desmedt A, English NJ. Hydrogen Inter-Cage Hopping and Cage Occupancies inside Hydrogen Hydrate: Molecular-Dynamics Analysis. Applied Sciences. 2021; 11(1):282. https://doi.org/10.3390/app11010282
Chicago/Turabian StyleKrishnan, Yogeshwaran, Mohammad Reza Ghaani, Arnaud Desmedt, and Niall J. English. 2021. "Hydrogen Inter-Cage Hopping and Cage Occupancies inside Hydrogen Hydrate: Molecular-Dynamics Analysis" Applied Sciences 11, no. 1: 282. https://doi.org/10.3390/app11010282