Abstract
Multimodal imaging gains increasing popularity for biomedical applications. This article presents the design of a novel multimodal imaging system. The centerpiece is a light microscope operating in the incident and transmitted light mode. Additionally, Raman spectroscopy and VIS/NIR reflectance spectroscopy are adapted. The proof-of-concept is realized to distinguish between grey matter (GM) and white matter (WM) of normal mouse brain tissue. Besides Raman and VIS/NIR spectroscopy, the following optical microscopy techniques are applied in the incident light mode: brightfield, darkfield, and polarization microscopy. To complement the study, brightfield images of a hematoxylin and eosin (H&E) stained cryosection in the transmitted light mode are recorded using the same imaging system. Data acquisition based on polarization microscopy and Raman spectroscopy gives the best results regarding the tissue differentiation of the unstained section. In addition to the discrimination of GM and WM, both modalities are suited to highlight differences in the density of myelinated axons. For Raman spectroscopy, this is achieved by calculating the sum of two intensity peak ratios (I2857 + I2888)/I2930 in the high-wavenumber region. For an optimum combination of the modalities, it is recommended to apply the molecule-specific but time-consuming Raman spectroscopy to smaller regions of interest, which have previously been identified by the microscopic modes.
1. Introduction
The gold standard for tissue examination is histological analysis [1]. In connection with disease, it is known as histopathology [2]. Especially since the inventions of the microtome for cutting thin tissue sections in 1839 and the hematoxylin and eosin (H&E) staining in 1875 [3], medical imaging using brightfield optical microscopy of stained cryosections is a well-established tool for research and diagnostic purposes [4]. This imaging technique is based on morphological differences within the tissue [4]. H&E as the standard dye promotes the identification of certain structures since acidophilic components such as cell cytoplasm, fiber structures, and interstitial stromal materials appear pale to deep pink, whereas basophilic components of the cell structure such as RNA and DNA are colored in blue [3,5]. Further morphological imaging techniques for biomedical applications are, for instance, optical coherence (OC) tomography [6,7,8,9], laser scanning microscopy [10,11,12], second-harmonic generation (SHG) microscopy [13,14], and two-photon excited fluorescence (TPEF) microscopy [15,16]. Those techniques have in common that they highlight structures within the tissue.
In contrast, functional imaging techniques visualize functional parameters such as the local chemical composition [4]. One of these imaging techniques is Raman-based microscopy. This technique is a combination of Raman spectroscopy and microscopy. It was first proposed by Delhaye and Dhamelincourt [17] in 1975 and has been commercially sold by companies since the 1990s [18]. Thereby, a microscope objective with a high numerical aperture (NA) focuses the excitation light to a small spot with a diameter of a few micrometers and enables a high collection efficiency of the Raman signal from a microscopic area [18]. The so-called spontaneous Raman effect was predicted by Adolf Smekal [19] in 1923 and experimentally confirmed by Raman and Krishnan [20] in 1928. Raman spectroscopy is an important vibrational spectroscopic method based on inelastic scattering of light by molecules. Hence, the frequencies of an incident photon, which interacts with a molecule, and the resulting scattered photon differ from each other. Raman scattered light with a lower frequency than the incident frequency is known as Stokes Raman scattering, and vice versa, anti-Stokes Raman scattering is Raman scattered light with a higher frequency. This frequency shift is indicative of discrete vibrational modes associated with chemical bonds within the molecule, independent of the wavelength of the excitation light source (laser). Consequently, qualitative measurements of chemical compositions can be obtained. Thus, Raman spectroscopy provides a label-free highly molecular specific technique. However, a change in polarizability of a chemical bonding orbital is crucial for the excitation of the fundamental vibration in a molecule and the observation of Raman spectra. Furthermore, Raman scattering is a weak effect with an occurrence probability of approximately 1 in 108 incident photons [21,22] compared to Rayleigh scattering where the light frequency remains constant (elastic scattering). In consequence, Raman spectroscopy requires intensive monochromatic light like lasers as a light source. The choice of laser wavelength and power plays an important role in Raman spectroscopy. The Raman intensity is linear to the laser’s power and proportional to the fourth power of the laser’s frequency [23]. This results in higher Raman signals for shorter excitation wavelengths.
In contrast, short laser wavelengths cause fluorescence signal that interferes with the Raman spectrum [24]. This is why an excitation wavelength in the red or NIR region is recommended for Raman spectroscopy of fluorescent biological material [23,25]. Another approach is the reduction of the detection volume with a confocal detection principle [18]. In this case, the image of a spot in the focal plane is detected through a pinhole in front of the detector [26]. Further fluorescence background in Raman microscopy is caused by standard glass microscope slides [27], the substrate for biological specimens. Investigations of aluminum foil, CaF2, quartz, low-emissivity, and Ag-coated and Au-coated microscope slides as potential substrates for Raman spectroscopy have been successfully performed [28,29]. Although the fluorescence signal can easily be six orders of magnitude higher than the Raman signal [18], which would result in an impossible identification of Raman peaks, taking the aforementioned factors into account, application of Raman-based microscopy for tissue analysis has been demonstrated multiple times.
The subsequent applications of Raman microscopy to classify tissue do not specify the Raman spectroscopy technique. Currently, more than 25 different types of known Raman techniques, including enhanced Raman spectroscopic techniques such as stimulated Raman scattering (SRS), coherent anti-Stokes Raman scattering (CARS) as well as surface- (SERS) and tip-enhanced (TERS) Raman scattering have emerged [30]. Raman microscopy was successfully applied by various researchers for ex-vivo examination of pathological changes of different tissue types such as breast [31,32], brain [33,34,35,36], colon [37], skin [38,39,40], and spinal cord [41,42]. Nearly all researchers prepared a histological stain (H&E staining) of the previously measured sample or a serial section to accompany an ex-vivo chemical image as it is an established method to visualize and analyze the morphology of the tissue [43]. The video capability to record the brightfield images of H&E-stained samples are often included in the Raman system. Besides the regular brightfield photographs, companies offer the integration of conventional microscopic modalities such as darkfield, polarized light, and epifluorescence within the Raman microscope [44].
Combinations of different modalities are referred to as multimodal imaging approaches [45]. One commonly used combination is Raman spectroscopy and infrared (IR) spectroscopy, which is an absorption-based vibrational spectroscopic technique. Kochan et al. [46] used Raman and IR imaging to study alteration in murine brain tissue. Reviews of tissue classifications with IR and/or Raman spectroscopy are published by Bunaciu et al. [47] and Lyng et al. [48]. Lasch and Noda [49] complemented Raman and IR spectroscopy by MALDI-TOF mass spectrometry to analyze hamster brain tissue. An overview of several tissue studies based on mass spectrometry and vibrational spectroscopy was recently given by Tuck et al. [43]. Combinations of Raman spectroscopy and the above-mentioned morphological imaging techniques have been developed as well. Bovenkamp et al. [50] and Egodage et al. [51] used OC tomography and Raman spectroscopy to analyze tissue. Bocklitz et al. [52] combined CARS, TPEF, and SHG to calculate a computational H&E stain for fast characterization of tissue architecture as well as the biochemical composition. Based on the calculated image, a region of interest (ROI) was determined for subsequent molecular selective diagnostic using Raman spectroscopy. If multiple measurement techniques are combined within one instrument, it is called a multimodal system [53].
To the best of the authors’ knowledge, the design of a multimodal imaging system combining brightfield, darkfield, and polarization microscopy, spontaneous Raman spectroscopy and VIS/NIR reflectance spectroscopy in the incident light mode as well as brightfield microscopy in the transmitted light mode have not been reported to distinguish grey matter (GM) and white matter (WM) of normal (under healthy conditions) brain tissue. GM and WM are two main areas within the brain that can be clearly differentiated [54]. GM is characterized by protein-rich neuronal bodies. WM does not contain neuronal bodies. The lipid-rich WM is characterized by a high presence of myelinated axons [54]. For Raman spectroscopy, this difference in lipid and protein content has been used to distinguish GM and WM based on intensity peak ratios and the sum of two intensity peak ratios by a series of researchers [33,34,35,46,55,56]. These lipid to protein ratios were used within this work to evaluate the detected Raman spectra. In total, two sets of measurements were performed for data acquisition. The first set of measurements was a global scan of the brain section. The second scan was a local scan of a smaller area. During both runs, all modalities were applied. This contains the incident light modes as well as the transmitted light mode for recording an H&E-stained sample. Thus, the presented multimodal imaging system is an all-in-one solution to measure unstained tissue samples with different modalities and complement these results by images of H&E-stained samples.
2. Materials and Methods
2.1. Sample Preparation
One female C57/Bl6J mouse (Janvier Labs, Le Genest-Saint-Isle, France) was perfused for 20 min with 1 × phosphate-buffered saline (PBS). Afterwards, the brain was removed, immediately embedded in optimal cutting temperature (OCT) compound (OCT Mounting media, VWR, Randor, PA, USA), and stored at −80 °C in order to avoid disintegration.
At a later time, a 16 µm thick section of the frozen brain was taken in the coronal plane by using a CM3050S cryostat (Leica, Wetzlar, Germany) at an operating temperature of −20 °C. This cryosection was mounted on an Au-coated glass slide (BioGold®, E63479-AS, Science Services GmbH, München, Germany). A consecutive 16 µm cryosection was collected on a SuperFrost® slide (VWR, Radnor, PA, USA) and stained with H&E for reference. While the H&E-stained tissue section was covered with Eukitt® mounting medium (Buddeberg GmbH, Mannheim, Germany) and a coverslip, the tissue section on the gold-coated slide was stored without embedding medium or coverslip at −20 °C until further analysis.
2.2. Multimodal Imaging System
The centerpiece of the multimodal imaging system (Figure 1) is the modular upright microscope Leica DM R (Leica Microsystems, Wetzlar, Germany) for transmitted and incident light microscopy. Table 1 specifies the numbered parts used throughout.
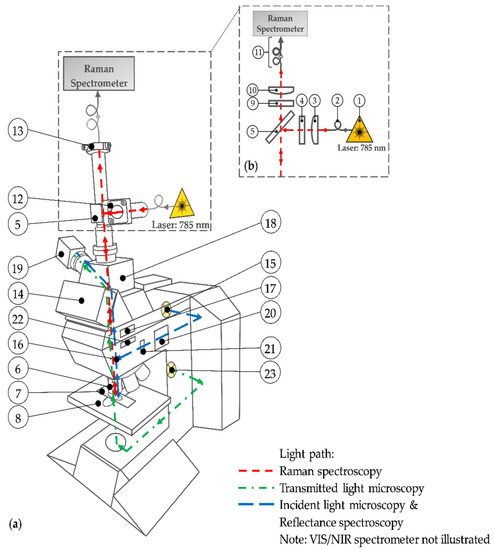
Figure 1.
(a) Schematic drawing of the multimodal imaging system; (b) light path of the expanded Raman spectroscopy. See Table 1 for the description of the used numbers.

Table 1.
Description of the numbering depicted in Figure 1.
Compared to the original device configuration, the setup is expanded by an additional light path (red path) for Raman spectroscopy (see Figure 1b). For this purpose, the commercially available MultiSpec® Raman System (tec5 AG, Steinbach, Germany) is used. The integrated temperature-stabilized 785 nm diode laser (1) with an adjustable output power between 50 mW and 500 mW is connected to the setup via a 100 µm (core diameter) optical fiber (2). A plano-convex lens (3) generates a collimated laser beam. The laser line bandpass filter (4) eliminates spectral noise by transmitting only the laser wavelength. Subsequently, a dichroic mirror (5) reflects the monochromatic light downwards at a 90° angle. After passing the microscope stand, the laser light is focused by a 50× objective (6) onto the sample. The tissue section is mounted on a microscope slide (7) and placed at the motorized microscope stage (8). The backscattered elastic and inelastic light is collected by the aforementioned microscope objective (6) and penetrates upwards through the microscope stand. The dichroic mirror (5) and the adjacent longpass filter (9) block the Rayleigh scattered laser light as well as the higher energetic anti-Stokes scattered light. Only the long-wave (Stokes) shifted light passes the filters and is focused by an achromatic lens (10) onto a 100 µm fiber. This fiber acts as a pinhole for the confocal setup and is coupled to a further 600 µm fiber (11) in order to achieve an entire illumination of the Raman spectrometer’s entrance slit. This spectrometer section is included in the MultiSpec® Raman System. Spectra are recorded using a thermoelectric (TE) cooled charge-coupled device (CCD) detector in a spectral range from 300 cm−1 to 3200 cm−1 with a spectral resolution of 7 cm−1. For maximum power of excitation light at the sample surface, a right-angle kinematic mirror mount with a dielectric mirror (12), not illustrated in Figure 1b, enables the adjustment of the laser beam and consequently an optimum illumination of the objective’s lens in the excitation path. In the detection path, a translating mount for the fiber adapter plate (13), not illustrated in Figure 1b, allows the lateral orientation of the pinhole fiber to maximize the detected Raman signal. Due to the weak Raman effect, the lenses in the Raman path contain an anti-reflective coating to reduce surface reflections. In addition, any optical component within the microscope stand, such as the tube optics, reflector or filter cubes, and beam splitters, is removed while performing Raman spectroscopy. The eyepieces of the trinocular (14) are removed permanently because of safety reasons. To obviate an undesired laser beam exit, the beam output is additionally covered. Since the observation path is disabled, the beam splitter of the trinocular is also permanently removed. Hence, the trinocular is effectively a connector within the whole setup.
A second light path for incident light microscopy is illustrated in blue. The light of a halogen lamp (15) propagates laterally along the incident light axis of the microscope. A brightfield reflector (16, not illustrated) reflects the light beam through either a 5× or a 20× objective (6) onto the sample. The backscattered light passes the reflector, which is basically a beam splitter, and the tube lens (17). The interchangeable photo adapter tube (18) with two clickstop positions for a switchable beam splitter (either 100% to the top (no optics) or 100% to the side) is activated to guide the light sidewise through a lens (unspecified) onto an 8-bit color (CCD) camera (19). For incident light brightfield microscopy, the diaphragm module (20) with the field diaphragm and the aperture diaphragm is activated by pushing in. The field diaphragm and the aperture diaphragm are disabled by pulling out. This is the case for incident light darkfield microscopy. Moreover, the reflector (16) is replaced by a darkfield reflector to generate the required darkfield illumination. The setup for incident light polarization microscopy is similar to the brightfield illumination path. However, a polarizer (21) in the excitation path and an analyzer (22) in the detection path is mandatory to perform polarization microscopy. In order to increase the polarization contrast, a whole-wave compensator (WWC) that permits extremely sensitive color contrast [57] can be activated by turning the analyzer upside down. Furthermore, a Smith reflector instead of the brightfield reflector is inserted for both polarization setups. Due to the objectives’ specifications, darkfield and polarization microscopy have to be performed with the 20× objective while incident light brigthfield microscopy is conducted with the 20× and the 5× objective. On the one hand, sample observation with the 5× objective facilitates the lateral orientation on the brain section because of larger image sections. On the other hand, as a result of similar resolution, the 5× objective allows the direct comparison of the photographs taken from the unstained sample with the photographs of the H&E-stained sample acquired by the transmitted light microscopy as described further down.
The blue light path is also used to perform reflectance spectroscopy in the visible (VIS) and near-infrared (NIR) region. Therefore, the camera is replaced by a multi-channel spectrometer (MCS) (not illustrated). In order to guide the light to the spectrometer, an additional plano-convex lens (LA1951, Thorlabs Inc., Newton, NJ, USA) is utilized to focus the beam onto a 50 µm pinhole fiber (M14L02) and an attached 550 µm fiber (FG550LEC) from Thorlabs Inc. (entire configuration not illustrated in Figure 1). The elastic scattered light between 310 nm and 950 nm is detected by the MCS 621 VIS II whereas the MCS 611 NIR 2.2 (both Carl Zeiss Spectroscopy GmbH, Jena, Germany) is used for the subsequent spectral region from 950 nm to 2150 nm. To cover the entire VIS/NIR spectral range, the IR blocking filter in the microscope’s lamp housing is removed.
The light of a second lower placed halogen lamp (23) propagates through the bottom part of the microscope stand (green path) and penetrates the sample from bottom to top. The light that transmits through the sample is collected by the 5× objective. After that, the light passes the tube lens (17), gets reflected at the switchable beam splitter (18), and is focused on the camera (19). This modality is called transmitted light microscopy. It is used to record photographs of the stained sample. Thus, the images of the unstained sample obtained with the aforementioned methods are complemented. The optical components in the excitation path of the transmitted light microscope are not illustrated Figure 1. However, they are implemented and adjusted according to the microscope’s manual [57].
2.3. Data Acquisition
The proof-of-concept consisted of two measurement sets (global and local) for tissue analysis, while one set of measurement comprised sequential tissue scans with each modality. These modalities, as already mentioned in Section 2.2, were subdivided into incident light modes (Figure 1, red and blue light path) to measure the unstained sample and the transmitted light mode (Figure 1, green light path) to record brightfield photographs of the H&E-stained sample (gold standard).
The measurements were started with the global scans in order to record the whole sample. Afterwards, local scans were carried out. The purpose of the local scans was to map ROI with smaller step sizes by achieving a higher spatial resolution of the hyperspectral data. The ROI was one tissue section with a boundary of GM (cortex) and WM (corpus callosum).
First of all, the frozen and unstained tissue section was left to thaw at room temperature. Measurements were also carried out at room temperature. No immersion medium between the sample surface and the microscope objectives was used. The microscope stage, the camera, as well as the spectrometers, were triggered by an in-house developed LabVIEW program (National InstrumentsTM) during the scanning procedure. In the present case, the sample on the microscope stage was scanned along the spatial axes (x- and y-axes) while either photographs or spectra were recorded at each measurement point. This methodology is called point mapping. Consequently, three-dimensional (x, y, z) data sets were acquired where x and y were the image coordinate and z was the spectrum or the photograph, respectively. The step size between two neighboring measurement points depended on the modality. The photographs were consistently taken with an overlap of approximately 20%. This resulted in a step size of 720 µm in the x-direction and 540 µm in y-direction for the 5× objective. With the 20× objective, samples were scanned in intervals of 180 µm along the x-axis and 135 µm along the y-axis. Raman spectra as well as VIS/NIR reflectance spectra were recorded pointwise with step sizes of either 200 µm (global scans) or 5 µm (local scans).
Prior to one set of measurement, a white balance of the camera was carried out in the incident light brightfield mode using the 5× objective and a Zenith® Diffuse Reflectance Standard (Reflectivity ≈ 99%, SG 3052, SphereOptics GmbH, Herrsching, Germany) as a reference. Therefore, the voltage value of the halogen lamp was set to 5 V and the camera’s integration time was 100 ms. Subsequently, data acquisition of the unstained tissue section was started with the incident light brightfield mode using the same operating voltage and magnification but at a shorter integration time of 10 ms. Those parameters were identical to the parameters of the following brightfield scan using the 20× objective.
The workflow of the data acquisition for one set of measurement, which includes several scanning runs, is shown in Table 2.
Table 2.
Workflow and setup parameters of one set of measurements for tissue analysis with the multimodal imaging system.
Due to a change in the illumination technique for darkfield microscopy and the weak signal of tissue for polarization microscopy, the light intensity was increased by maximizing the lamp’s voltage value (12 V) for the mentioned modalities. The integration time was adjusted so that neither an underexposure nor an overexposure of the photographs was recognized. The exact values are shown in Table 2.
Afterwards, Raman map measurements were performed. The laser power was set to 100% resulting in 57 mW at the sample surface. Each spectrum was recorded for 4000 ms at a single accumulation. In advance, a dark signal measurement without laser light was executed to detect photon counts caused by the detector. This offset signal is a function of temperature and exposure time [58]. It was removed from each Raman spectrum in order to achieve raw spectra. Dark signals at the switched-off halogen lamp were also recorded for VIS and NIR spectroscopy and subtracted from the measured spectra via the LabVIEW program because of the same reason. The operating voltage of the halogen lamp and the integration time for the associated modality are indicated Table 2. The number of accumulations for VIS and NIR spectra acquisition was set to 1. An additional scan (*, Table 2 was performed after Raman spectroscopy and before VIS spectroscopy that did not contribute further local tissue information. This second scan with the brightfield modality (operating parameters similar to scan no. 2) was a good indicator to determine the tissue condition. The comparison of the results from the initial brightfield scan and the post Raman brightfield scan could indicate visible photodamage caused by the laser.
The last step (scan no. 9) within one set of measurements was the recording of the H&E-stained sample on the glass slide with the transmitted light operating mode. Measurements were carried out with the 5× objective and at a voltage value of the halogen lamp of 12 V. The exposure time per photograph was set to 10 ms. In contrast to the incident light microscopy modes, the white balance was performed with the open-source raster graphics editor GIMP after stitching.
2.4. Image Creation and Spectra Processing
The analysis of the data was executed separately for each modality. Photographs taken by the camera were stitched to an entire image using the Grid/Collection stitching [59], a plug-in for the open-source image processing package Fiji (Fiji Is Just ImageJ). No further processing of the stitched images like contrast enhancement or brightness adjustment was performed. An in-house developed Python3 (Python Software Foundation) application assembled hyperspectral data cubes for the respective spectroscopic modalities. Such data cubes assign each recorded spectrum to the corresponding image coordinate (pixel). Before spectral analysis, preprocessing of the raw spectra was required to eliminate undesirable spectral features. In the case of VIS/NIR reflectance spectroscopy, the spectral characteristics of the halogen lamp had to be eliminated by an appropriate reference measurement. In order to get spectral information of the sample reflectivity, reflection spectra R(λ) of each pixel were calculated in the VIS spectral region as well as the NIR spectral region. Therefore, Equation (1) [60] was used.
where is the measured intensity reflected from the sample as a function of wavelength and is a reference spectrum. As the reference spectrum, the intensity spectrum of the halogen lamp reflected from the aforementioned Zenith® Diffuse Reflectance Standard was used after multiplication by a factor of 2.5. This arbitrary factor was applied to raise the intensity level of the diffuse reflection by the Zenith® standard above the intensity level of sample measurements caused by the specular reflection of the gold-coated microscope slide.
In order to create an image based on the VIS or NIR spectra, the reflectivity at a certain wavelength was visualized by colorizing the pixels according to their value. Therefore, PyQtGraph (Scientific Graphics and GUI Library for Python) and the linear color scale bipolar was used. The highest value was indicated by a yellow-colored pixel while the pixel with the lowest value was light blue. The same color scale was used to create images based on the Raman spectra. In contrast to the data analysis of the VIS/NIR reflection spectra, intensity peak ratios of two Raman peaks were calculated to distinguish between GM and WM. To get these desired Raman peaks, preprocessing of the Raman spectra was mandatory. Spectral preprocessing of Raman spectra was investigated by several researchers in the past [58,61,62]. In this publication, Raman spectra were preprocessed as follows. First of all, the spectral range was limited to the range from 350 cm−1 to 3195 cm−1 in order to delete software-based zero values that would cause errors in the further processing steps. As a consequence, the number of x-values (active sensor pixels) was reduced from originally 1044 to 955. Cosmic rays that appear randomly when CCD detectors were struck from high energy particles were removed by applying an algorithm for spectra despiking [63]. The threshold value was set to 65. Afterwards, the Raman spectra’s offset was removed by subtracting the Raman spectrum’s lowest intensity value from each sensor pixel. With the aid of an asymmetric least squares smoothing, initially developed by Eilers in 2005 and presented by He et al. [64] in 2014, broad background from tissue fluorescence was compensated. Within the applied algorithm, the lambda value and the p-value were set to 1,000,000 and 0.1, respectively. The number of iterations was set to 10. Data preprocessing was completed by smoothing the Raman spectra in order to reduce noise. Therefore, a Savitzky–Golay filter [65] with a 3rd order polynomial fit and a window size of 11 was applied. The complete spectral preprocessing, as well as image creation based on spectral data, was performed with the developed Python3 application.
3. Results
Figure 2 presents the stitched images obtained with the 5× objective. Column 1 shows images of the H&E-stained sample recorded with the transmitted light brightfield mode. The incident light brightfield mode was used to take photographs of the unstained tissue section (column 2). The stitched images are the results from scan number 1 (incident light mode) and 9 (transmitted light mode) within the measurement sets for tissue analysis (Table 2).
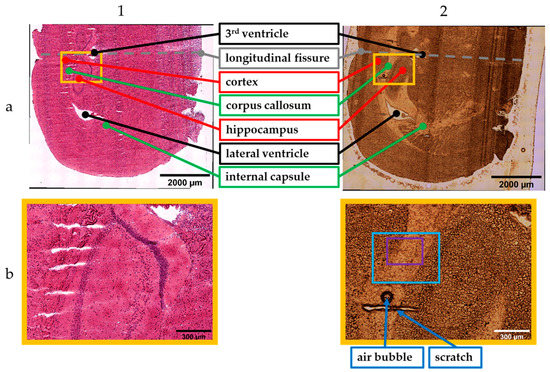
Figure 2.
Stitched images (5× objective) of the stained (1) and unstained (2) sample. Results of (a) global and (b) local scan; photographs: 10 × 12 (1a), 11 × 12 (2a), 2 × 2 (1b,2b) (see text for details).
Since the brain was cut in the coronal plane, the tissue section showed symmetric brain structures that were mirrored on the longitudinal fissure (dashed line, Figure 2(1a,2a)). Consequently, it was sufficient to measure one cerebral hemisphere for the global scans. The global scan’s result of the H&E-stained sample is illustrated in Figure 2(1a). The corresponding scan of the unstained sample is shown in Figure 2(2a). Both images provide an overall overview of the cryosections.
The main parts within the tissue sections were examined by experienced researchers trained in mouse anatomy and regions of interest were identified and marked accordingly (see Figure 2(1a,2a)). The annotations are in accordance with the labelling in the Allen Brain Atlas [66,67].Besides GM and WM, ventricles can be observed. These are cavities filled with cerebrospinal fluid [68]. The lateral and the 3rd ventricle are highlighted in black. The corpus callosum as well as the internal capsule are WM. Those regions are marked in green. GM, the hippocampus and the cortex, is highlighted in red.
Figure 2(1b,2b) show smaller image sections containing portions of the cortex, corpus callosum, and hippocampus. These stitched images are the results of the local scan using the 5× magnification. These smaller areas can be localized by the orange frame within the global images (Figure 2(1a,2a)). Furthermore, artefacts such as an air bubble and a scratch facilitate the visual assignment within the unstained tissue section (Figure 2(2b)).
Although the stitched images of the unstained sample allow a visual differentiation from GM and WM, staining is necessary to highlight cellular structure. The cell density of the WM is lower compared to GM. This fact is confirmed by the images of the H&E-stained tissue section that show an accumulation of the dark colored cells in the GM.
The area of the local scan using the microscopic modes and the 20× objective is highlighted by the bright blue frame (see Figure 2(2b)). Spectra acquisition with the molecule-specific but time-consuming Raman spectroscopy and the VIS/NIR spectroscopy was performed within the smaller ROI (purple frame). The total measuring times, including instrumentation time (tstage, treadout, ttransfer), resulted in 12 min for VIS spectra and 16 min for NIR spectra. In contrast, the total time to acquire Raman spectra was nearly 3 hrs. Taking photographs took less than 1 min for each modality.
The total measuring time varied from 30 min for incident light brightfield microscopy to 90 min for Raman spectroscopy. The respective integration times and step sizes are listed in Table 2.
The subsequent evaluation (Figure 3) displays images of the unstained tissue section’s brain structure based on the different modalities. These are the results of the global scan and the local scan obtained during scan number 2–8 within one set of measurements.
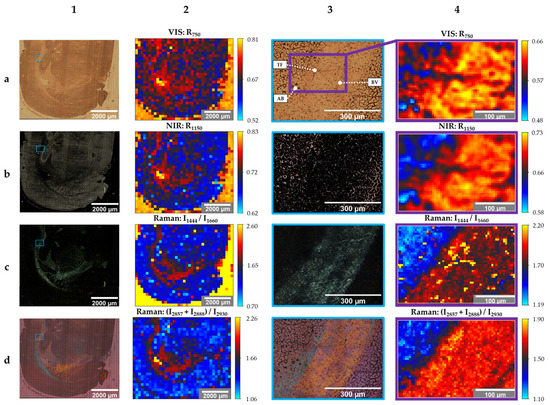
Figure 3.
Stitched images (20× objective, column 1 + 3) and created images (50× objective, column 2 + 4) of the unstained sample; modes: brightfield (1a,3a), darkfield (1b,3b), polarization (1c,1d,3c,3d), reflectance spectroscopy (2a,2b,4a,4b), Raman spectroscopy (2c,2d,4c,4d); photographs: 41 × 45 (1a–d), 3 × 3 (3a–d); spectra: 37 × 31 (2a–d), 61 × 41 (4a–d); travel range: 7200 µm × 7200 µm (1a–d, 2a–d), scanning area: 584 µm × 438 µm (3a–d), 305 µm × 205 µm (4a–d) abbreviations: AB: air bubbles, BV: blood vessel, TF: tissue fold (see text for details).
Columns 1 and 2 show the images resulted from the global scan. The local scan’s results are shown in columns 3 and 4. The columns with even numbers contain created images based on the evaluation of spectra, while column number 1 and 3 show stitched photographs. Information regarding the number of photographs or spectra and the travel range or scanning area of each image are provided in the figure caption of Figure 3.
Image 1a of Figure 3 is the brightfield-microscopic image of the unstained sample’s cerebral hemisphere. In comparison to Figure 2(2a), photographs were taken with the 20× objective and a higher resolution, consequently. Figure 3(1b) is the stitched image of the same tissue section, obtained with the darkfield mode. While darkfield microscopy highlights GM bright, the same morphological structure appears dark in brightfield images.
Another modality that highlights WM is polarized light microscopy. WM shines bright in Figure 3(1c), while the other parts of the image are dark. A qualitative color-coded differentiation of the WM is obtained by polarization microscopy with the activated WWC (Figure 3(1d)). While the internal capsule is highlighted in orange, the majority of the corpus callosum is colored in blue. The bright green segment (red circle) at the lower right part of the tissue, which is also visible in the previously mentioned images, could be identified as an artefact that does not belong to the brain section.
The local scan’s stitched images in column 3 (Figure 3(3a–d)) display the area of the blue frame in column 1 (Figure 3(1a–d)) in an enlarged image.
The smaller area within Figure 3(3a) (purple frame) represents the local scan’s area, where spectra were acquired. This smaller ROI comprises a boundary between cortex (GM) and corpus callosum (WM). Moreover, three landmarks for further data evaluation were determined. These include three air bubbles (AB) in the lower-left corner, a tissue fold (TF) in the center, and a blood vessel (BV) in the lower-right corner.
Especially the air bubbles and the tissue fold can be identified in the created images based on reflectance spectra in the VIS (Figure 3(4a)) and NIR (Figure 3(4b)) region. Figure 3(4a) shows the reflectivity of the tissue sample at 750 nm (R750). The qualitative reflectance spectra in the VIS region are illustrated in Figure 4a.
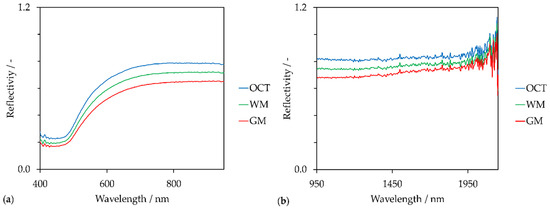
Figure 4.
Reflectance spectra (averaging over 9 spectra) of white matter (WM), grey matter (GM) and optimal cutting temperature compound (OCT) in the (a) VIS spectral region, (b) NIR spectral region.
Besides the reflectance spectra of GM and WM, the reflectance spectrum of OCT is shown as well. Since the tissue was embedded in transparent OCT, OCT still surrounds the cryosection and consequently coats the microscope slide (see Figure 2(2a)). Although OCT is a side component within this proof-of-concept, OCT affects the spectral evaluation of the global scans. This is shown in Figure 3(2a). Compared to the few pixels located at the lateral ventricle and the single pixel of the 3rd ventricle that are yellow, the OCT area is rather orange. This confirms that OCT reduces the intensity of the backscattered light compared to the reflectivity of the gold-coated slide without tissue sample. However, reflectivity at OCT is higher than the reflectivity at WM and GM. Due to the 180° back-scattering arrangement, the incident light transmits the sample twice. Forward-scattered light is reflected back by the coated surface. Gold absorbs in the blue spectral range. This is indicated by the drop around 450 nm in each reflectance spectrum of Figure 4a. Reflectivity increases between 500 nm and 700 nm and remains constant from 700 nm to 950 nm. The highest reflectivity of the shown spectra is detected for OCT. GM has the lowest reflectivity in the VIS spectral region. This is in accordance with the color-coding in Figure 3(2a,4a). Pixels in the WM area appear reddish and GM is rather bluish. Artefacts, such as the three air bubbles and the tissue fold, that appear darker in the brightfield images have lower reflectivity values as well.
However, the created images (Figure 3(2a,4a)) are based on reflectivity at 750 nm. The chosen wavelength lies in the optical window of tissue that ranges from 600 nm to 940 nm [69]. This spectral region is independent of the main absorber in blood-perfused tissue, hemoglobin [70]. Therefore, the created images do not contain information regarding the chemical composition of the tissue section but represent the reflectivity of the sample at 750 nm.
Similar results were obtained by images based on the reflectivity in the NIR spectral region at 1150 nm (R1150) (Figure 3(2b,4b)). This wavelength lies also in an optical window around 1100 nm to 1350 nm [71]. Compared to the VIS spectra, NIR spectra fluctuate across the entire wavelength region (Figure 4b). This could be a result of the NIR spectrometer’s lower sensor sensibility (extended wavelength InGaAs detector) compared to the VIS spectrometer (Si photodiodes). The signal-to-noise ratio becomes poorer with increasing wavelengths. Anyhow, these fluctuations could be the reason for poorer contrast and lateral resolution of the NIR images compared to the VIS images.
Images of the tissue section that are based on chemical composition were obtained by the evaluation of the Raman spectra. As already mentioned in the introduction, intensity peak ratios as well as the sum of two intensity peak ratios of lipid and protein peaks have been identified to distinguish GM and WM. Representative Raman spectra of WM, GM, and OCT are shown in Figure 5. Peaks used to create Raman images are highlighted within the figure. The corresponding assignments of intensity peaks to lipid and protein are indicated in Table 3.
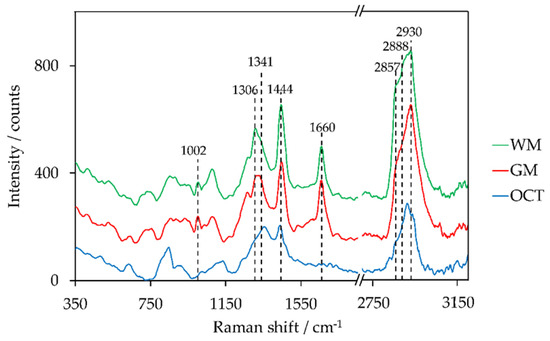
Figure 5.
Preprocessed Raman spectra (averaging over 9 spectra) of white matter (WM), grey matter (GM), and optimal cutting temperature compound (OCT), offset: +150 counts (GM), +300 counts (WM).
Table 3.
Raman peak position and assignment of functional groups to distinguish GM and WM.
The majority of the peaks are located in the fingerprint region between 700 cm−1 and 1800 cm−1 [75]. The high-wavenumber region (2800–3050 cm−1) contains CH-stretching vibrations [76]. The evaluated peak ratios of Raman intensities to distinguish GM and WM were adapted from literature and include the following: I1299/I1439 [55], I1330/I1344 [35], I1441/I1659 [34], I1442/I1662 [33], I1443/I1008 [46], I2845/I2930 [56], (I2857 + I2888)/I2930 [46].
The best results to distinguish GM, WM, and OCT within the global scan were obtained with an intensity peak ratio of I1444/I1660 (Figure 3(2c)), where 1444 cm−1 is primarily assigned to lipids (CH2, CH3 vibration modes) and 1660 cm−1 corresponds to proteins (Amide I band). The highest peak ratios were achieved for OCT (yellow pixels). This can be explained by the fact that the Raman spectrum of OCT does not show a peak around 1660 cm−1. The peak ratio is higher in WM than in GM. This is confirmed by the peak characteristics within the shown Raman spectra. The ventricles do not show color contrast. The reason, therefore, is the broad fluorescence background of the glass signal at approximately 1400 cm−1 when using 785-nm laser excitation [27]. The glass signal originates from the glass microscope slide that is coated by a 120 nm thin gold film. This background signal was not completely removed within the data preprocessing. The corresponding intensity peak ratios of the local scan (Figure 3(4c)) show a clear boundary between GM (blue) and WM (red). The yellow pixels confirm the suspicion of a tissue fold within WM since an overlay in lipid-rich tissue results in an increase of the intensity peak ratio. The blood vessel is emphasized by the blue pixels due to the lower peak ratio.
The sum of two Raman intensity peak ratios (I2857 + I2888)/I2930 in the high-wavenumber range enables the distinction between GM and WM in the local ROI (Figure 3(4d)). Besides the clear boundary between the two brain regions, the blood vessel can be easily identified. Furthermore, the yellow pixels within the WM correspond to the obtained blue pigmentation within the corpus callosum in Figure 3(3d) in case of polarization microscopy with WWC. In contrast to the evaluation method I1444/I1660, a sufficient differentiation of GM and OCT based on the CH stretching modes of the Raman spectra was not possible (Figure 3(2d)). However, the evaluation method (I2857 + I2888)/I2930 highlighted the ventricles and the WM within the global scan.
4. Discussion
The scanning results show that different information content is provided based on the modality. Brightfield and darkfield images are complementary techniques to visualize the morphological structure of the unstained tissue. The image obtained from the evaluation of the local scan’s reflectance spectra in the VIS spectral region can be visually localized within the brightfield image of the local scan. However, the spatial resolution of the microscopic image is higher than the spatial resolution of the spectroscopic-based image. While the step size for spectra acquisition of the local scan is 5 µm, the pixel size of a microscopic photograph using the 20× magnification is 0.17 µm. In contrast, common color cameras are based on three color channels (red, green, blue), whereas spectrometer record hyperspectral data where each intensity value is assigned to a wavelength. The calculated VIS reflectance spectra did not show absorption bands other than the absorption of the gold coating. The main absorber in the tissue below 600 nm is hemoglobin. Since neither the camera detected a reddish area nor the spectra showed the hemoglobin-specific reflectivity drops between 500 and 600 nm, it can be assumed that hemoglobin was no longer present in the tissue section due to a successful perfusion of the animal. Another indicator that confirms the quality of sample preparation was the absence of a Raman peak around 1585 cm−1 that could be assigned to hemorrhage [34].
Similar to the reflectance spectra in the VIS wavelength range, NIR reflectance spectra did not contain molecular-specific information. Confocal imaging of deep mouse brain with long-wavelength (NIR) illumination was demonstrated by Xia et al. [77]. The researchers took advantage of the tissue’s lower light absorption and scattering in the optical window around 1700 nm. An excitation light source in this NIR region combined with reflectance confocal microscopy enabled deep penetration into tissue with high spatial resolution. The results were images at various depths based on reflectivity in the NIR region. A prominent absorber in the NIR region is water with absorption bands at 1450 nm and 1900 nm [78]. Weak NIR absorption bands within tissue originate from biomolecules such as lipids, collagen, and proteins. Because of the biomolecules, poor light absorption in the NIR spectral range a sample thickness of 16 µm was not sufficient to detect molecule-specific information in the NIR range. Additionally, absorption bands of these biomolecules are usually overlapped by the strong absorption properties of water [78]. A statement regarding the air-dried tissue’s water content based on other modalities of the imaging system is not possible.
The detection range of the Raman spectrometer does not cover the high-wavenumber range between 3200 cm−1 and 3500 cm−1. Otherwise, the detection of the OH bonds’ vibrations near 3245 cm−1 and 3425 cm−1 would be possible. Despite the polarity of the water molecule, those Raman peaks are primarily assigned to water [79]. Moreover, Kirsch et al. [79] recognized lower water content in WM compared to GM of normal brain. This caused Kochan et al. [46] to suggest that variation in tissue thickness during tissue drying could falsify the results. Due to higher water content, GM would become thinner than WM. As a consequence of this height difference, higher intensity values would be recorded within WM areas [46]. To exclude that this assumption applies to the presented results, the height profile of a tissue section containing the smaller ROI of the local scan was determined using the digital microscope VHX-7000 (Keyence Deutschland GmbH, Neu-Isenburg, Germany), see Figure A1 (Appendix A). The color map does not show a correlation between the height and the tissue type. Therefore, the determined reflectivity within the VIS and NIR spectral range is a characteristic of the tissue itself rather than its physical property. For the same reason, the chemical information based on Raman spectroscopy is rated as reliable.
The color coding within the created Raman images (Figure 3(2c–d,4c–d)) confirms the observations of Mizuno et al. [33], Kast et al. [34], and Kochan et al. [46], who detected the higher intensity peak ratio and the sum of intensity peak ratios within the WM. The peak positions of the measured Raman peaks within WM and GM were in good accordance with the peak positions assigned in the literature (see Table 3). A significant shift of the Raman peak positions is observed for OCT. Below 1400 cm−1, a shift to higher wavenumbers was determined, and vice versa, a shift to lower wavenumbers occurred above 1400 cm−1. This is an indicator for the detection of the broad fluorescence background of the glass slide. Since this trend in shift was not recognized within the Raman spectra of GM and WM, it is assumed that the laser light is primarily scattered and absorbed by the optical properties of the tissue. However, a prolongation of the laser beam and a subsequent decrease in depth resolution due to the mismatch of refractive indices was studied by Everall et al. [80,81,82,83]. The replacement of the dry objective by a water immersion objective for Raman measurements would be a possibility to increase the depth resolution and to reduce the background signal. Bonnier et al. [39] analyzed human skin by Raman spectroscopy at various excitation wavelengths with a water immersion objective. Besides the reduction of the background signal, Bonnier et al. observed a decrease in photodamage at lower laser wavelength due to thermal heat sink. Anyhow, a switch in immersion medium might result in an adaption of the Raman’s excitation light path because of variation in the pupil’s lens diameter for different objectives.
Nevertheless, the discrimination of GM and WM based on the differences of the chemical compositions was possible with the presented Raman path. Especially the sum of two peak ratios in the high-wavenumber region led to good results for the local scan. The high values that were indicated by the yellow and reddish pixels can be explained by previous observations of Mizuno et al. [33]. Already in 1992, he reported an increase in the peaks at 2852 cm−1 and 2885 cm−1 in the WM and myelin fraction, whereas the peak at 2938 cm−1 was high in all brain tissues. In 2018, Ji et al. [84] identified the CH stretch region as an indicator to visualize the myelinated axons. Especially the peak around 2850 cm−1, which is assigned to the CH2 stretch modes of lipids. Since the location of the yellow pixels within the WM of the local scan were in accordance with the blue pigmentation in the polarization image with the activated WWC, the blue color suggests a higher density of myelinated axons. Lower densities, which are primarily present in the internal capsule, were indicated in orange. The polarization microscopy without the WWC did not show a color difference of the various structure within WM. Additionally, this modality required a three times longer acquisition time. Therefore, polarization microscopy with WWC is preferable.
The gold-coated microscope slide enhanced the contrast in brightfield images. Another modality that profits from the reflective gold coating is Raman spectroscopy where the excitation laser passes through the tissue twice. The selected integration time of 4 s was sufficient to detect proper Raman spectra. Moreover, the integration time did not result in visual photodamage of the tissue. This was confirmed by calculating the mean-squared error based on a pixel-to-pixel comparison of two images. Therefore, the local scan’s brightfield image obtained within step no. 2 and the additional brightfield image after the Raman scan (step no. *) were used (see Appendix A, Figure A2). The comparison resulted in a mean-squared error of 0.05. Although the maximum recommended accumulation time of 10 s [39] to conduct a tissue scan in a realistic time was not extended, it is advised that the spectroscopic modalities are exclusively applied to scan designated ROIs. One reason is the long measuring time per data point, the other reason is the spatial resolution. Although all spectroscopic modalities were able to distinguish GM and WM within the global scans, the mapping area was under-sampled at step sizes of 200 µm; moreover, the OCT interfered with the evaluation. Much better are the created images of the local scan. In all images, it can clearly be distinguished between GM and WM. The chosen step size of 5 µm was experimentally determined as the smallest step size of the microscope stage.
As expected, the images of the H&E-stained sample did not show any abnormalities regarding the cellular structure of the brain sample. For further studies, the brightfield microscopic image of the H&E-stained sample could be used to determine an ROI. In order to compare the H&E-stained sample one-to-one with the different microscopic images of an unstained sample, images should have similar pixel resolution. Recording photographs of the H&E-stained sample with the 20× magnification and the multimodal imaging system is possible without any problems. Only the activation of the condenser in the excitation path and the adaption of the illumination (Koehler illumination) are required. This opens up new possibilities for the multimodal imaging system to include the results of the H&E-stained sample in the multimodal data evaluation for tissue research and diagnostic within ex-vivo applications. Those applications are not limited to the separation of GM and WM although structural changes within GM and/or WM are indicative for patients with HIV [85], multiple sclerosis [86], and Alzheimer’s disease [87,88]. Ex-vivo applications of any tissue section for research and diagnostic purposes are possible.
The expansion of further modalities, such as epifluorescence microscopy, is feasible without any problems. This is due to the modular principle of the microscope. Space for an extra lamp housing and filters is already provided in the incident light as well as the transmitted light path of the modular microscope.
5. Conclusions
The presented multimodal imaging system was successfully applied to distinguish GM and WM of ex-vivo mouse brain tissue. The brightfield images of the H&E-stained sample highlighted differences in the cellular structure of the tissue. Brightfield and darkfield images emphasized the brains morphology by complementary imaging modes. Although the information of the spectral reflectivity was exclusively based on surface characteristics, VIS spectroscopy seems to be a promising tool to receive hyperspectral data to calculate tissue absorptions, if blood is present. Molecular-specific information regarding tissue components was achieved by Raman spectroscopy. The best result regarding the distinction of GM, WM, and OCT within the global scan was achieved by an intensity peak ratio in the fingerprint region (I1444/I1660). Using this evaluation method GM, WM, as well as tissue structures like a tissue fold and a blood vessel, were highlighted within the local scan. The discrimination of the blood vessel, GM and WM was also possible with a second evaluation method (I2857 + I2888)/I2930. Additionally, this evaluation method enabled the identification of differences regarding the density of myelinated axons within WM. The created image was in good accordance with the image obtained with polarization microscopy (active WWC). For further applications, it is recommended to use microscopic-based images to get an overview and determine ROIs. A combination of the image obtained from the H&E-stained sample and the various images of the unstained sample is recommended. The spectroscopic modalities, especially the molecular specific but time-consuming Raman spectroscopy, should be applied to designated areas to create images of the tissue’s molecular composition.
Author Contributions
Conceptualization, A.H., T.B., and M.R.; methodology, A.H., J.D., and L.S.; software, S.S. and F.W.; validation, A.H. and T.B.; investigation, A.H. and J.D; resources, J.D., L.S., and M.R.; data curation, S.S. and F.W; writing—original draft preparation, A.H. and T.B.; writing—review and editing, S.S., F.W., J.D., L.S., and M.R.; visualization, A.H. and F.W.; supervision, A.H., T.B., and M.R.; project administration, A.H.; funding acquisition, M.R. All authors have read and agreed to the published version of the manuscript.
Funding
The article processing charge was funded by the Baden-Württemberg Ministry of Science, Research and Culture as part of the open-access publication fund. L.S. was supported by intramural funding provided by the Medical Faculty Mannheim of Heidelberg University and a research grant from the Hertie Foundation (medMS MyLab, P1180016).
Institutional Review Board Statement
The C57/Bl6J mouse was purchased from Janvier labs (Le Genest-Saint-Isle, France). The mouse was housed under specific pathogen-free conditions at the animal facility Mannheim and kept on a 12 hr light/dark cycle. Food and water were provided ad libitum. The animal experimental protocol was approved by the local animal ethics committee (I-20/11).
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A

Figure A1.
Color map including height scale of an unstained tissue section’s height profile (magnification: 500×). Purple rectangle frames the area of the smaller ROI recorded with the spectroscopic modalities (see Figure 3). Abbreviations: AB: air bubbles, BV: blood vessel, TF: tissue fold (see text for details).
Figure A1.
Color map including height scale of an unstained tissue section’s height profile (magnification: 500×). Purple rectangle frames the area of the smaller ROI recorded with the spectroscopic modalities (see Figure 3). Abbreviations: AB: air bubbles, BV: blood vessel, TF: tissue fold (see text for details).


Figure A2.
Stitched images (20× objective) of the unstained sample modes. Results of (a) step no. 2 (identical to Figure 3(3a)) and (b) step no. * of the local scan.
Figure A2.
Stitched images (20× objective) of the unstained sample modes. Results of (a) step no. 2 (identical to Figure 3(3a)) and (b) step no. * of the local scan.

References
- Paramitha, D.; Ulum, M.; Purnama, A.; Wicaksono, D.; Noviana, D.; Hermawan, H. Monitoring degradation products and metal ions in vivo. In Monitoring and Evaluation of Biomaterials and their Performance In Vivo; Narayan, R., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 19–38. [Google Scholar]
- Krafft, C.; Jürgen, P. Vibrational Spectroscopic Imaging of Soft Tissue. In Infrared and Raman Spectroscopic Imaging; 2nd Completely Revised and Updated Edition; Salzer, R., Siesler, H.W., Eds.; Wiley-VCH: Weinheim, Germany, 2014; pp. 113–152. ISBN 978-3-527-33652-4. [Google Scholar]
- Mireskandari, M.; Petersen, I. Clinical Pathology. In Ex-vivo and In-vivo Optical Molecular Pathology; Popp, J., Ed.; Wiley: Weinheim, Germany, 2014; pp. 1–26. ISBN 978-3-527-33513-8. [Google Scholar]
- Vogler, N.; Heuke, S.; Bocklitz, T.W.; Schmitt, M.; Popp, J. Multimodal Imaging Spectroscopy of Tissue. Annu. Rev. Anal. Chem. (Palo Alto California) 2015, 8, 359–387. [Google Scholar] [CrossRef]
- Schäfer, L. Histologische Techniken. In Histopathologie der Haut; Cerroni, L., Garbe, C., Metze, D., Kutzner, H., Kerl, H., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 3–13. ISBN 978-3-662-45132-8. [Google Scholar]
- Baumann, B. Optical Coherence Tomotgraphy for Brain Imaging. In Advanced Optical Methods for Brain Imaging; Kao, F.-J., Keiser, G., Gogoi, A., Eds.; Springer: Singapore, 2019; pp. 49–68. ISBN 978-981-10-9019-6. [Google Scholar]
- Pfister, M.; Schützenberger, K.; Schäfer, B.J.; Puchner, S.; Stegmann, H.; Hohenadl, C.; Mildner, M.; Garhöfer, G.; Schmetterer, L.; Werkmeister, R.M. Optical Coherence Tomography Angiography Monitors Cutaneous Wound Healing under Angio-genesis-Promoting Treatment in Diabetic and Non-Diabetic Mice. Appl. Sci. 2021, 11, 2447. [Google Scholar] [CrossRef]
- Liu, G.; Chen, Z. Optical Coherence Tomography for Brain Imaging. In Optical Methods and Instrumentation in Brain Imaging and Therapy; Madsen, S.J., Ed.; Springer: New York, NY, USA, 2013; pp. 157–172. ISBN 978-1-4614-4977-5. [Google Scholar]
- Zysk, A.M.; Boppart, S.A. Optical Coherence Tomography. In Optical Imaging and Microscopy: Techniques and Advanced Systems; 2nd Revised Edition; Török, P., Kao, F.-J., Eds.; Springer: Berlin, Germany, 2007; pp. 401–436. ISBN 978-3-540-69563-9. [Google Scholar]
- Guthoff, R.F.; Baudouin, C.; Stave, J. Atlas of Confocal Laser Scanning In-Vivo Microscopy in Ophthalmology: Principles and Applications in Diagnostic and Therapeutic Ophthalmology; Springer: Berlin, Germany, 2006; ISBN 978-3-540-32705-9. [Google Scholar]
- Hofmann-Wellenhof, R.; Pellacani, G.; Malvehy, J.; Soyer, H.P. Reflectance Confocal Microscopy for Skin Diseases; Springer: Berlin, Germany, 2012; ISBN 978-3-642-21996-2. [Google Scholar]
- Bağcı, I.S.; Aoki, R.; Krammer, S.; Ruzicka, T.; Sárdy, M.; Hartmann, D. Ex vivo confocal laser scanning microscopy: An in-novative method for direct immunofluorescence of cutaneous vasculitis. J. Biophotonics 2019, 12, e201800425. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, M.; Quinn, K.P.; Kobler, J.B.; Zeitels, S.M.; Georgakoudi, I.; Ben-Yakar, A. Quantitative differentiation of normal and scarred tissues using second-harmonic generation microscopy. Scanning 2016, 38, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.-Y.; Chang, Y.-L.; Lo, W.; Hsueh, C.-M.; Chen, W.-L.; Ghazaryan, A.A.; Hu, P.-S.; Young, T.-H.; Chen, S.-J.; Dong, C.-Y. Characterizing the morphologic changes in collagen crosslinked-treated corneas by Fourier transform-second harmonic generation imaging. J. Cataract Refract. Surg. 2013, 39, 779–788. [Google Scholar] [CrossRef]
- Kawakami, R.; Nemoto, T. In Vivo Imaging of All Cortical Layers and Hippocampal CA1 Pyramidal Cells by Two-Photon Excitation Microscopy. In Advanced Optical Methods for Brain Imaging; Kao, F.-J., Keiser, G., Gogoi, A., Eds.; Springer: Singapore, 2019; pp. 113–122. ISBN 978-981-10-9019-6. [Google Scholar]
- Piston, D.W. Two-Photon Excitation Microscopy for Three-Dimensional Imaging of Living Intact Tissues. In Fluorescence Microscopy: From Principles to Biological Applications, Second Edition; Kubitscheck, U., Ed.; Wiley-VCH: Weinheim, Germany, 2017; pp. 203–242. ISBN 978-3-527-33837-5. [Google Scholar]
- Delhaye, M.; Dhamelincourt, P. Raman microprobe and microscope with laser excitation. J. Raman Spectrosc. 1975, 3, 33–43. [Google Scholar] [CrossRef]
- Hollricher, O. Raman Instrumentation for Confocal Raman Microscopy. In Confocal Raman Microscopy; Dieing, T., Ed.; Springer: Berlin, Germany, 2010; pp. 43–60. ISBN 978-3-642-12522-5. [Google Scholar]
- Smekal, A. Zur Quantentheorie der Dispersion. Naturwissenschaften 1923, 11, 873–875. [Google Scholar] [CrossRef]
- Raman, C.V.; Krishnan, K.S. A New Type of Secondary Radiation. Nature 1928, 121, 501–502. [Google Scholar] [CrossRef]
- Butler, H.J.; Ashton, L.; Bird, B.; Cinque, G.; Curtis, K.; Dorney, J.; Esmonde-White, K.; Fullwood, N.J.; Gardner, B.; Martin-Hirsch, P.L.; et al. Using Raman spectroscopy to characterize biological materials. Nat. Protoc. 2016, 11, 664–687. [Google Scholar] [CrossRef]
- Petry, R.; Schmitt, M.; Popp, J. Raman spectroscopy--a prospective tool in the life sciences. Chemphyschem 2003, 4, 14–30. [Google Scholar] [CrossRef]
- Kiselev, R.; Schie, I.W.; Aškrabić, S.; Krafft, C.; Popp, J. Design and first applications of a flexible Raman micro-spectroscopic system for biological imaging. BSI 2016, 5, 115–127. [Google Scholar] [CrossRef]
- Vandenabeele, P. Practical Raman Spectroscopy: An Introduction; Wiley: Chichester, UK, 2013; ISBN 978-0-470-68319-4. [Google Scholar]
- Schalk, R.; Heintz, A.; Braun, F.; Iacono, G.; Rädle, M.; Gretz, N.; Methner, F.-J.; Beuermann, T. Comparison of Raman and Mid-Infrared Spectroscopy for Real-Time Monitoring of Yeast Fermentations: A Proof-of-Concept for Multi-Channel Photometric Sensors. Appl. Sci. 2019, 9, 2472. [Google Scholar] [CrossRef]
- Hollricher, O.; Ibach, W. High-Resolution Optical and Confocal Microscopy. In Confocal Raman Microscopy; Dieing, T., Ed.; Springer: Berlin, Germany, 2010; pp. 1–20. ISBN 978-3-642-12522-5. [Google Scholar]
- Tuschel, D. Selecting an Excitation Wavelength for Raman Spectroscopy. Spectroscopy 2016, 31, 14–23. [Google Scholar]
- Cui, L.; Butler, H.J.; Martin-Hirsch, P.L.; Martin, F.L. Aluminium foil as a potential substrate for ATR-FTIR, transflection FTIR or Raman spectrochemical analysis of biological specimens. Anal. Methods 2016, 8, 481–487. [Google Scholar] [CrossRef]
- Fullwood, L.M.; Griffiths, D.; Ashton, K.; Dawson, T.; Lea, R.W.; Davis, C.; Bonnier, F.; Byrne, H.J.; Baker, M.J. Effect of sub-strate choice and tissue type on tissue preparation for spectral histopathology by Raman microspectroscopy. Analyst 2014, 139, 446–454. [Google Scholar] [CrossRef]
- Jones, R.R.; Hooper, D.C.; Zhang, L.; Wolverson, D.; Valev, V.K. Raman Techniques: Fundamentals and Frontiers. Nanoscale Res. Lett. 2019, 14, 231. [Google Scholar] [CrossRef] [PubMed]
- Brozek-Pluska, B.; Kopec, M. Raman microspectroscopy of Hematoporphyrins. Imaging of the noncancerous and the cancerous human breast tissues with photosensitizers. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2016, 169, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Abramczyk, H.; Brozek-Pluska, B.; Kopec, M. Polarized Raman microscopy imaging: Capabilities and challenges for cancer research. J. Mol. Liq. 2018, 259, 102–111. [Google Scholar] [CrossRef]
- Mizuno, A.; Hayashi, T.; Tashibu, K.; Maraishi, S.; Kawauchi, K.; Ozaki, Y. Near-infrared FT-Raman spectra of the rat brain tissues. Neurosci. Lett. 1992, 141, 47–52. [Google Scholar] [CrossRef]
- Kast, R.E.; Auner, G.W.; Rosenblum, M.L.; Mikkelsen, T.; Yurgelevic, S.M.; Raghunathan, A.; Poisson, L.M.; Kalkanis, S.N. Raman molecular imaging of brain frozen tissue sections. J. Neurooncol. 2014, 120, 55–62. [Google Scholar] [CrossRef]
- Kast, R.; Auner, G.; Yurgelevic, S.; Broadbent, B.; Raghunathan, A.; Poisson, L.M.; Mikkelsen, T.; Rosenblum, M.L.; Kalkanis, S.N. Identification of regions of normal grey matter and white matter from pathologic glioblastoma and necrosis in frozen sections using Raman imaging. J. Neurooncol. 2015, 125, 287–295. [Google Scholar] [CrossRef]
- Kalkanis, S.N.; Kast, R.E.; Rosenblum, M.L.; Mikkelsen, T.; Yurgelevic, S.M.; Nelson, K.M.; Raghunathan, A.; Poisson, L.M.; Auner, G.W. Raman spectroscopy to distinguish grey matter, necrosis, and glioblastoma multiforme in frozen tissue sections. J. Neurooncol. 2014, 116, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Brozek-Pluska, B.; Musial, J.; Kordek, R.; Abramczyk, H. Analysis of Human Colon by Raman Spectroscopy and Imaging-Elucidation of Biochemical Changes in Carcinogenesis. Int. J. Mol. Sci. 2019, 20, 3398. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.M.; Bonnier, F.; Tfayli, A.; Lambkin, H.; Flynn, K.; McDonagh, V.; Healy, C.; Clive Lee, T.; Lyng, F.M.; Byrne, H.J. Raman spectroscopic analysis of human skin tissue sections ex-vivo: Evaluation of the effects of tissue processing and dewaxing. J. Biomed. Opt. 2013, 18, 61202. [Google Scholar] [CrossRef] [PubMed]
- Bonnier, F.; Ali, S.M.; Knief, P.; Lambkin, H.; Flynn, K.; McDonagh, V.; Healy, C.; Lee, T.C.; Lyng, F.M.; Byrne, H.J. Analysis of human skin tissue by Raman microspectroscopy: Dealing with the background. Vib. Spectrosc. 2012, 61, 124–132. [Google Scholar] [CrossRef]
- Bonnier, F.; Mehmood, A.; Knief, P.; Meade, A.D.; Hornebeck, W.; Lambkin, H.; Flynn, K.; McDonagh, V.; Healy, C.; Lee, T.C.; et al. In vitro analysis of immersed human tissues by Raman microspectroscopy. J. Raman Spectrosc. 2011, 42, 888–896. [Google Scholar] [CrossRef]
- Wang, S.; Liang, Z.; Gong, Y.; Yin, Y.; Wang, K.; He, Q.; Wang, Z.; Bai, J. Confocal raman microspectral imaging of ex vivo human spinal cord tissue. J. Photochem. Photobiol. B 2016, 163, 177–184. [Google Scholar] [CrossRef]
- Li, J.; Liang, Z.; Wang, S.; Wang, Z.; Zhang, X.; Hu, X.; Wang, K.; He, Q.; Bai, J. Study on the pathological and biomedical characteristics of spinal cord injury by confocal Raman microspectral imaging. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2019, 210, 148–158. [Google Scholar] [CrossRef]
- Tuck, M.; Blanc, L.; Touti, R.; Patterson, N.H.; van Nuffel, S.; Villette, S.; Taveau, J.-C.; Römpp, A.; Brunelle, A.; Lecomte, S.; et al. Multimodal Imaging Based on Vibrational Spectroscopies and Mass Spectrometry Imaging Applied to Biological Tissue: A Multiscale and Multiomics Review. Anal. Chem. 2021, 93, 445–477. [Google Scholar] [CrossRef]
- De Bettignies, P. Optics/instrumentation. Phys. Sci. Rev. 2020, 5. [Google Scholar] [CrossRef]
- Bashkatov, A.N.; Zakharov, V.P.; Bucharskaya, A.B.; Borisova, E.G.; Khristoforova, Y.A.; Genina, E.A.; Tuchin, V.V. Malignant Tissue Optical Properties. In Multimodal Optical Diagnostics of Cancer; Tuchin, V.V., Popp, J., Zakharov, V., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 3–43. ISBN 978-3-030-44593-5. [Google Scholar]
- Kochan, K.; Chrabaszcz, K.; Szczur, B.; Maslak, E.; Dybas, J.; Marzec, K.M. IR and Raman imaging of murine brains from control and ApoE/LDLR(−/−) mice with advanced atherosclerosis. Analyst 2016, 141, 5329–5338. [Google Scholar] [CrossRef] [PubMed]
- Bunaciu, A.A.; Hoang, V.D.; Aboul-Enein, H.Y. Vibrational Micro-Spectroscopy of Human Tissues Analysis: Review. Crit. Rev. Anal. Chem. 2017, 47, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Lyng, F.; Ramos, I.; Ibrahim, O.; Byrne, H. Vibrational Microspectroscopy for Cancer Screening. Appl. Sci. 2015, 5, 23–35. [Google Scholar] [CrossRef]
- Lasch, P.; Noda, I. Two-Dimensional Correlation Spectroscopy for Multimodal Analysis of FT-IR, Raman, and MALDI-TOF MS Hyperspectral Images with Hamster Brain Tissue. Anal. Chem. 2017, 89, 5008–5016. [Google Scholar] [CrossRef] [PubMed]
- Bovenkamp, D.; Sentosa, R.; Rank, E.; Erkkilä, M.; Placzek, F.; Püls, J.; Drexler, W.; Leitgeb, R.; Garstka, N.; Shariat, S.; et al. Combination of High-Resolution Optical Coherence Tomography and Raman Spectroscopy for Improved Staging and Grading in Bladder Cancer. Appl. Sci. 2018, 8, 2371. [Google Scholar] [CrossRef]
- Egodage, K.; Dochow, S.; Bocklitz, T.; Chernavskaia, O.; Matthaeus, C.; Schmitt, M.; Popp, J. The combination of optical co-herence tomography and Raman spectroscopy for tissue characterization. JBPE 2015, 169–177. [Google Scholar] [CrossRef]
- Bocklitz, T.W.; Salah, F.S.; Vogler, N.; Heuke, S.; Chernavskaia, O.; Schmidt, C.; Waldner, M.J.; Greten, F.R.; Bräuer, R.; Schmitt, M.; et al. Pseudo-HE images derived from CARS/TPEF/SHG multimodal imaging in combination with Raman-spectroscopy as a pathological screening tool. BMC Cancer 2016, 16, 534. [Google Scholar] [CrossRef]
- Das, N.K.; Dai, Y.; Liu, P.; Hu, C.; Tong, L.; Chen, X.; Smith, Z.J. Raman Plus X: Biomedical Applications of Multimodal Raman Spectroscopy. Sensors 2017, 17, 1592. [Google Scholar] [CrossRef]
- Sanchez-Molina, P.; Kreuzer, M.; Benseny-Cases, N.; Valente, T.; Almolda, B.; González, B.; Castellano, B.; Perálvarez-Marín, A. From Mouse to Human: Comparative Analysis between Grey and White Matter by Synchrotron-Fourier Transformed Infrared Microspectroscopy. Biomolecules 2020, 10, 1099. [Google Scholar] [CrossRef]
- Beljebbar, A.; Dukic, S.; Amharref, N.; Manfait, M. Ex vivo and in vivo diagnosis of C6 glioblastoma development by Ra-man spectroscopy coupled to a microprobe. Anal. Bioanal. Chem. 2010, 398, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.; Orringer, D.A.; Freudiger, C.W.; Ramkissoon, S.; Liu, X.; Lau, D.; Golby, A.J.; Norton, I.; Hayashi, M.; Agar, N.Y.R.; et al. Rapid, label-free detection of brain tumors with stimulated Raman scattering microscopy. Sci. Transl. Med. 2013, 5, 201ra119. [Google Scholar] [CrossRef]
- Leica Microsystems Wetzlar GmbH. Leica DM R: Instructions; Leica Microsystems: Wetzlar, Germany, 2000. [Google Scholar]
- Zoubir, A. Raman Imaging: Techniques and Applications; Springer: Berlin/Heidelberg, Germany, 2012; ISBN 978-3-642-28251-5. [Google Scholar]
- Preibisch, S.; Saalfeld, S.; Tomancak, P. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics 2009, 25, 1463–1465. [Google Scholar] [CrossRef] [PubMed]
- Otto, M. Analytische Chemie; VCH: Weinheim, Germany, 1995; ISBN 9783527286911. [Google Scholar]
- Byrne, H.J.; Knief, P.; Keating, M.E.; Bonnier, F. Spectral pre and post processing for infrared and Raman spectroscopy of biological tissues and cells. Chem. Soc. Rev. 2016, 45, 1865–1878. [Google Scholar] [CrossRef] [PubMed]
- Bocklitz, T.; Walter, A.; Hartmann, K.; Rösch, P.; Popp, J. How to pre-process Raman spectra for reliable and stable models? Anal. Chim. Acta 2011, 704, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, D.; Hayes, K. A Simple Algorithm for Despiking Raman Spectra. Chemom. Intell. Lab. Syst. 2018, 179, 82–84. [Google Scholar] [CrossRef]
- He, S.; Zhang, W.; Liu, L.; Huang, Y.; He, J.; Xie, W.; Wu, P.; Du, C. Baseline correction for Raman spectra using an improved asymmetric least squares method. Anal. Methods 2014, 6, 4402–4407. [Google Scholar] [CrossRef]
- Savitzky, A.; Golay, M.J.E. Smoothing and Differentiation of Data by Simplified Least Squares Procedures. Anal. Chem. 1964, 36, 1627–1639. [Google Scholar] [CrossRef]
- © 2004 Allen Institute for Brain Science. Allen Mouse Brain Atlas. Available online: https://mouse.brain-map.org/experiment/thumbnails/100048576?image_type=atlas (accessed on 15 May 2021).
- Lein, E.S.; Hawrylycz, M.J.; Ao, N.; Ayres, M.; Bensinger, A.; Bernard, A.; Boe, A.F.; Boguski, M.S.; Brockway, K.S.; Byrnes, E.J.; et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007, 445, 168–176. [Google Scholar] [CrossRef]
- Schröder, H.; Moser, N.; Huggenberger, S. Neuroanatomy of the Mouse; Springer International Publishing: Cham, Switzerland, 2020; ISBN 978-3-030-19897-8. [Google Scholar]
- Braun, F.; Schalk, R.; Heintz, A.; Feike, P.; Firmowski, S.; Beuermann, T.; Methner, F.-J.; Kränzlin, B.; Gretz, N.; Rädle, M. NADH-fluorescence scattering correction for absolute concentration determination in a liquid tissue phantom using a novel multispectral magnetic-resonance-imaging-compatible needle probe. Meas. Sci. Technol. 2017, 28, 75903. [Google Scholar] [CrossRef]
- Sikurova, L.; Balis, P.; Zvarik, M. Penetration of laser light through red blood cell ghosts. J. Photochem. Photobiol. B 2011, 103, 230–233. [Google Scholar] [CrossRef]
- Golovynskyi, S.; Golovynska, I.; Stepanova, L.I.; Datsenko, O.I.; Liu, L.; Qu, J.; Ohulchanskyy, T.Y. Optical windows for head tissues in near-infrared and short-wave infrared regions: Approaching transcranial light applications. J. Biophotonics 2018, 11, e201800141. [Google Scholar] [CrossRef] [PubMed]
- Shim, M.G.; Wilson, B.C. The effects of ex vivo handling procedures on the near-infrared Raman spectra of normal mamma-lian tissues. Photochem. Photobiol. 1996, 63, 662–671. [Google Scholar] [CrossRef]
- Ong, C.W.; Shen, Z.X.; He, Y.; Lee, T.; Tang, S.H. Raman microspectroscopy of the brain tissues in the substantia nigra and MPTP-induced Parkinson’s disease. J. Raman Spectrosc. 1999, 30, 91–96. [Google Scholar] [CrossRef]
- Dybas, J.; Marzec, K.M.; Pacia, M.Z.; Kochan, K.; Czamara, K.; Chrabaszcz, K.; Staniszewska-Slezak, E.; Malek, K.; Baranska, M.; Kaczor, A. Raman spectroscopy as a sensitive probe of soft tissue composition-Imaging of cross-sections of various organs vs. single spectra of tissue homogenates. TrAC Trends Anal. Chem. 2016, 85, 117–127. [Google Scholar] [CrossRef]
- Diem, M.; Mazur, A.; Lenau, K.; Schubert, J.; Bird, B.; Miljković, M.; Krafft, C.; Popp, J. Molecular pathology via IR and Raman spectral imaging. J. Biophotonics 2013, 6, 855–886. [Google Scholar] [CrossRef] [PubMed]
- Aubertin, K.; Desroches, J.; Jermyn, M.; Trinh, V.Q.; Saad, F.; Trudel, D.; Leblond, F. Combining high wavenumber and fin-gerprint Raman spectroscopy for the detection of prostate cancer during radical prostatectomy. Biomed. Opt. Express 2018, 9, 4294–4305. [Google Scholar] [CrossRef] [PubMed]
- Xia, F.; Wu, C.; Sinefeld, D.; Li, B.; Qin, Y.; Xu, C. In vivo label-free confocal imaging of the deep mouse brain with long-wavelength illumination. Biomed. Opt. Express 2018, 9, 6545–6555. [Google Scholar] [CrossRef]
- Sanchez-Cano, A.; Saldaña-Díaz, J.E.; Perdices, L.; Pinilla, I.; Salgado-Remacha, F.J.; Jarabo, S. Measurement method of optical properties of ex vivo biological tissues of rats in the near-infrared range. Appl. Opt. 2020, 59, D111–D117. [Google Scholar] [CrossRef]
- Kirsch, M.; Schackert, G.; Salzer, R.; Krafft, C. Raman spectroscopic imaging for in vivo detection of cerebral brain metastases. Anal. Bioanal. Chem. 2010, 398, 1707–1713. [Google Scholar] [CrossRef]
- Everall, N.J. Confocal Raman Microscopy: Why the Depth Resolution and Spatial Accuracy Can Be Much Worse Than You Think. Appl. Spectrosc. 2000, 54, 1515–1520. [Google Scholar] [CrossRef]
- Everall, N.; Lapham, J.; Adar, F.; Whitley, A.; Lee, E.; Mamedov, S. Optimizing depth resolution in confocal Raman micros-copy: A comparison of metallurgical, dry corrected, and oil immersion objectives. Appl. Spectrosc. 2007, 61, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Everall, N.J. Confocal Raman microscopy: Performance, pitfalls, and best practice. Appl. Spectrosc. 2009, 63, 245A–262A. [Google Scholar] [CrossRef] [PubMed]
- Everall, N.J. Confocal Raman microscopy: Common errors and artefacts. Analyst 2010, 135, 2512–2522. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.; Arbel, M.; Zhang, L.; Freudiger, C.W.; Hou, S.S.; Lin, D.; Yang, X.; Bacskai, B.J.; Xie, X.S. Label-free imaging of amyloid plaques in Alzheimer’s disease with stimulated Raman scattering microscopy. Sci. Adv. 2018, 4, eaat7715. [Google Scholar] [CrossRef] [PubMed]
- Küper, M.; Rabe, K.; Esser, S.; Gizewski, E.R.; Husstedt, I.W.; Maschke, M.; Obermann, M. Structural grey and white matter changes in patients with HIV. J. Neurol. 2011, 258, 1066–1075. [Google Scholar] [CrossRef]
- Singh, S.; Tench, C.R.; Tanasescu, R.; Constantinescu, C.S. Localised Grey Matter Atrophy in Multiple Sclerosis and Clinically Isolated Syndrome-A Coordinate-Based Meta-Analysis, Meta-Analysis of Networks, and Meta-Regression of Voxel-Based Morphometry Studies. Brain Sci. 2020, 10, 798. [Google Scholar] [CrossRef]
- Lama, R.K.; Lee, S.-W. White Matter Network Alterations in Alzheimer’s Disease Patients. Appl. Sci. 2020, 10, 919. [Google Scholar] [CrossRef]
- Michael, R.; Lenferink, A.; Vrensen, G.F.J.M.; Gelpi, E.; Barraquer, R.I.; Otto, C. Hyperspectral Raman imaging of neuritic plaques and neurofibrillary tangles in brain tissue from Alzheimer’s disease patients. Sci. Rep. 2017, 7, 15603. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).