
Early Performance Prediction in Bioinformatics Systems Using Palladio Component Modeling


Abstract
:1. Introduction
2. Background
2.1. Software Performance Engineering (SPE)
- Measurement-based performance evaluation: in this technique, the values of the important performance requirements are collected by processing and analyzing collected data. A. Van Hoorn stated that the usual measurement-based operations to operate and improve the software are debugging, profiling, logging, and monitoring. Debugging and profiling generally occur at development time in the development environment, where a high degree of disruption is suitable. Logging and monitoring are used during operation in the production environment [16].
- Model-based performance evaluation: here, the values of performance measures of interest are obtained by analyzing and simulating the software system. Model-based predictions search for other alternatives concerning the deployment and architecture of a system. It can be useful in many ways, such as the prediction and comparison of the performance metrics according to the design alternatives introduced for the system by improving its architecture, and also producing early estimates of the performance measures of interest during the SDLC [17].
2.2. Palladio Component Modeling (PCM)
2.3. Bioinformatics Systems
3. Materials and Methods
3.1. Genome Comparison and Phylogenetic Analysis System
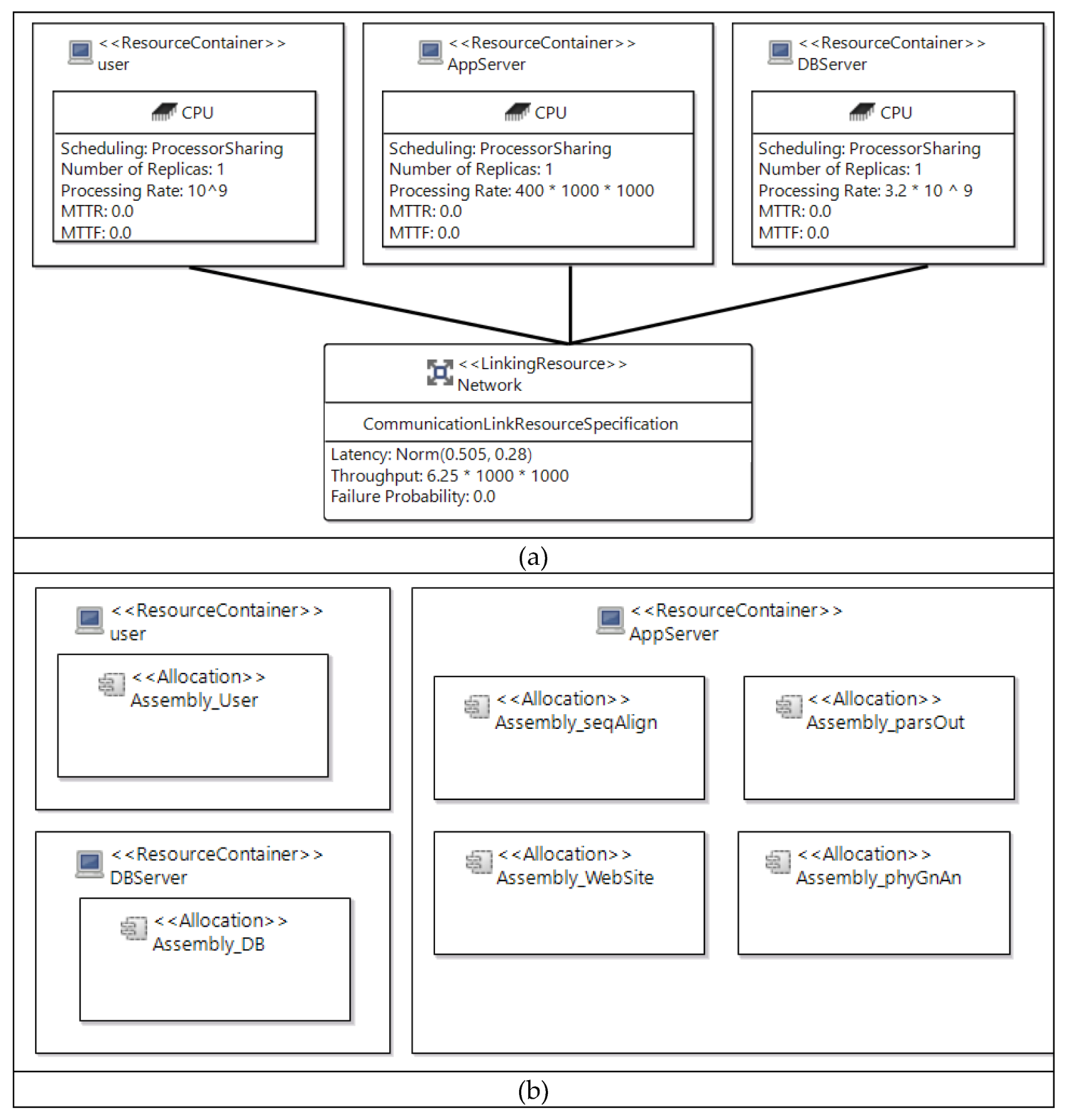
3.2. Performance Modeling and Simulation Using PCM
3.3. Proposed Alternative Design
4. Results
5. Discussion
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Smith, C.U.; Williams, L.G. Software performance engineering. In Encyclopedia of Software Engineering; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2003; pp. 794–810. [Google Scholar]
- Verma, K.K.; Solanki, A.K. A Novel Performance Analysis Technique Using Modeling and Refactoring for Software Architecture. In Proceedings of the International Conference on Advances in Engineering Science Management & Technology (ICAESMT), Uttaranchal University, Dehradun, India, 14–15 March 2019. [Google Scholar]
- Woodside, M.; Franks, G.; Petriu, D.C. The Future of Software Performance Engineering. In Proceedings of the FOSE’07, Minneapolis, MN, USA, 23–25 May 2007; pp. 171–187. [Google Scholar]
- NCBI. Available online: https://www.ncbi.nlm.nih.gov/books/NBK44939/ (accessed on 30 April 2021).
- Xia, X. Comparative genomics. In Handbook of Statistical Bioinformatics; Springer: Berlin/Heidelberg, Germany, 2011; pp. 567–600. [Google Scholar]
- Jarvis, P.D.; Holl, B.R.; Sumner, J.G. Phylogenetic invariants and Markov invariants. In Reference Module in Life Sciences; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Ashley, E. Towards precision medicine. Nat. Rev. Genet. 2016, 17, 507–522. [Google Scholar] [CrossRef]
- Huang, W.; Umbach, D.M.; Li, L. Accurate anchoring alignment of divergent sequences. Bioinformatics 2006, 22, 29–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, D.A.; Bergman, C.M.; Stoye, J.; Celniker, S.E.; Eisen, M.B. Benchmarking tools for the alignment of functional noncoding DNA. BMC Bioinform. 2004, 5, 6. [Google Scholar]
- Okonechnikov, K.; Golosova, O.; Fursov, M. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [PubMed] [Green Version]
- Negoita, G.A. High Performance Computing Applications: Inter-Process Communication, Workflow Optimization, and Deep Learning for Computational Nuclear Physics. Ph.D. Thesis, Iowa State University, Ames, IA, USA, 2018. [Google Scholar]
- Ayres, D.L.; Cummings, M.P.; Baele, G.; Darling, A.E.; Lewis, P.O.; Swofford, D.L.; Huelsenbeck, J.P.; Lemey, P.; Rambaut, A.; Suchard, M.A. BEAGLE 3: Improved Performance, Scaling, and Usability for a High-Performance Computing Library for Statistical Phylogenetics. Syst. Biol. 2019, 68, 1052–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabregat, A.; Sidiropoulos, K.; Viteri, G.; Forner, O.; Marin-Garcia, P.; Arnau, V.; D’Eustachio, P.; Stein, L.; Hermjakob, H. Reactome pathway analysis: A high-performance in-memory approach. BMC Bioinform. 2017, 18, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shajii, A.; Numanagić, I.; Baghdadi, R.; Berger, B.; Amarasinghe, S. Seq: A high-performance language for bioinformatics. Proc. ACM Program. Lang. 2019, 3, 1–29. [Google Scholar] [CrossRef] [Green Version]
- Cortellessa, V.; Di Marco, A.; Inverardi, P. Model-Based Software Performance Analysis; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Van Hoorn, A. Model-Driven Online Capacity Management for Component-Based Software Systems. Ph.D. Thesis, Kiel University, Kiel, Germany, 2014. [Google Scholar]
- Balsamo, S.; Marco, A.; Inverardi, P. Model-based performance prediction in software development: A survey. IEEE Trans. Softw. Eng. 2004, 30, 295–310. [Google Scholar] [CrossRef]
- Liao, L.; Chen, J.; Li, H.; Zeng, Y.; Shang, W.; Guo, J.; Sporea, C.; Toma, A.; Sajedi, S. Using black-box performance models to detect performance regressions under varying workloads: An empirical study. Empir. Softw. Eng. 2020, 25, 4130–4160. [Google Scholar] [CrossRef]
- Bertolino, A.; Inverardi, P.; Muccini, H. Software architecture-based analysis and testing: A look into achievements and future challenges. Computing 2013, 95, 633–648. [Google Scholar] [CrossRef]
- Object Management Group (OMG). UML-SPT: UML Profile for Schedulability, Performance, and Time, v 1.1. January 2005. Available online: https://www.omg.org/spec/SPTP/1.1/About-SPTP/ (accessed on 20 February 2021).
- Mallet, F.; André, C.; DeAntoni, J. Executing AADL Models with UML/MARTE. In Proceedings of the 14th IEEE International Conference on Engineering of Complex Computer Systems, Potsdam, Germany, 2–4 June 2009; pp. 371–376. [Google Scholar]
- Becker, S.; Koziolek, H.; Reussner, R. The Palladio Component Model for Model-driven Performance Prediction. J. Syst. Softw. 2009, 82, 3–22. [Google Scholar] [CrossRef]
- Ortega-Arjona, J.L.; Roberts, G. Architectural Performance Models: Estimating the Contribution of Software Structure to the Performance of Parallel Software Architecture. In Proceedings of the 2nd Nordic Workshop on Software Architecture, Ronneby, Sweden, 12–13 August 1999. [Google Scholar]
- Di Marco, A.; Mirandola, R. Model Transformation in Software Performance Engineering. In Proceedings of the International Conference on the Quality of Software Architectures, Västerås, Sweden, 27–29 June 2006; Hofmeister, C., Crnkovic, I., Reussner, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 95–110. [Google Scholar]
- Li, C.; Altamimi, T.; Zargar, M.; Casale, G.; Petriu, D. Tulsa: A Tool for Transforming UML to Layered Queueing Networks for Performance Analysis of Data Intensive Applications. In Proceedings of the International Conference on Quantitative Evaluation of Systems, Berlin, Germany, 5–7 September 2017; pp. 295–299. [Google Scholar]
- Brosig, F.; Meier, P.; Becker, S.; Koziolek, A.; Koziolek, H.; Kounev, S. Quantitative Evaluation of Model-Driven Performance Analysis and Simulation of Component-based Architectures. IEEE Trans. Softw. Eng. 2015, 41, 157–175. [Google Scholar] [CrossRef] [Green Version]
- Bolch, G.; Greiner, S.; De Meer, H.; Trivedi, K.S. Queueing Networks and Markov Chains: Modeling and Performance Evaluation with Computer Science Applications, 2nd ed.; Wiley and Sons: Hoboken, NJ, USA, 2006. [Google Scholar]
- Bernardi, S.; Donatelli, S.; Merseguer, J. From UML Sequence Diagrams and Statecharts to Analysable Petri Net Models. In Proceedings of the International Workshop on Software and Performance (WOSP), Rome, Italy, 24–26 July 2002; pp. 35–45. [Google Scholar]
- Koziolek, H. Performance evaluation of component-based software systems: A survey. Perform. Eval. 2010, 67, 634–658. [Google Scholar] [CrossRef] [Green Version]
- Trubiani, C.; Koziolek, A. Detection and solution of software performance antipatterns in palladio architectural models. In Proceedings of the International Conference on Performance Engineering (ICPE), Karlsruhe, Germany, 14–16 March 2011; pp. 19–30. [Google Scholar]
- Cortellessa, V.; Di Marco, A.; Eramo, R.; Pierantonio, A.; Trubiani, C. Digging into UML models to remove performance antipatterns. In Proceedings of the International Conference on Software Engineering, Cape Town, South Africa, 3 May 2010; pp. 9–16. [Google Scholar]
- Cortellessa, V.; Di Marco, A.; Trubiani, C. Software performance antipatterns: Modeling and analysis. In Formal Methods for Model-Driven Engineering; Bernardo, M., Cortellessa, V., Pierantonio, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 7320, pp. 290–335. [Google Scholar]
- Becker, S.; Koziolek, H.; Reussner, R. Model-based performance prediction with the palladio component model. In Proceedings of the 6th International Workshop on Software and Performance (WOSP2007), Buenos Aires, Argentina, 5–8 February 2007. [Google Scholar]
- Reussner, R.; Becker, S.; Burger, E.; Happe, J.; Hauck, M.; Koziolek, A.; Koziolek, H.; Krogmann, K.; Kuperberg, M. The Palladio Component Model; Karlsruhe Institute of Technology: Karlsruhe, Germany, 2011. [Google Scholar]
- Lawlor, B.; Walsh, P. Engineering bioinformatics: Building reliability, performance and productivity into bioinformatics software. Bioengineered 2015, 6, 193–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J. Bioinformatics, an introduction for computer scientists. ACM Comput. Surv. 2004, 36, 122–158. [Google Scholar] [CrossRef]
- Field, M.A. Detecting pathogenic variants in autoimmune diseases using high-throughput sequencing. Immunol. Cell Biol. 2020, 99, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.H.; Bogusz, M.; Whelan, S. Identifying Clusters of High Confidence Homologies in Multiple Sequence Alignments. Mol. Biol. Evol. 2019, 36, 2340–2351. [Google Scholar] [CrossRef] [PubMed]
- BLAST. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastSearch&BLAST_SPEC=blast2seq&LINK_LOC=align2seq (accessed on 10 October 2020).
- Clustal Omega. Available online: https://www.ebi.ac.uk/Tools/msa/clustalo/# (accessed on 10 October 2020).
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genome Comparison and Phylogenetic Analysis System. Available online: https://genomecomparison.wixsite.com/gecphans (accessed on 12 October 2020).
- Pezoa, F.; Reutter, J.L.; Suarez, F.; Ugarte, M.; Vrgoc, D. Foundations of JSON schema. In Proceedings of the 25th International Conference on World Wide Web, Montreal, QC, Canada, 11–15 April 2016; pp. 263–273. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UML Object | PCM Component |
|---|---|
| User 1 | user |
| Website 1 | webSite |
| Alignment Job 2 | seqAlignJob |
| Phylogenetic analysis Job 2 | PhyGnAnJob |
| Server 1 | parseOut |
| DB Manager 1 | DB |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dorgham, D.M.T.; Belal, N.A.; Abdelmoez, W. Early Performance Prediction in Bioinformatics Systems Using Palladio Component Modeling. Appl. Sci. 2021, 11, 5426. https://doi.org/10.3390/app11125426
Dorgham DMT, Belal NA, Abdelmoez W. Early Performance Prediction in Bioinformatics Systems Using Palladio Component Modeling. Applied Sciences. 2021; 11(12):5426. https://doi.org/10.3390/app11125426
Chicago/Turabian StyleDorgham, Doaa M. Talaat, Nahla A. Belal, and Walid Abdelmoez. 2021. "Early Performance Prediction in Bioinformatics Systems Using Palladio Component Modeling" Applied Sciences 11, no. 12: 5426. https://doi.org/10.3390/app11125426
APA StyleDorgham, D. M. T., Belal, N. A., & Abdelmoez, W. (2021). Early Performance Prediction in Bioinformatics Systems Using Palladio Component Modeling. Applied Sciences, 11(12), 5426. https://doi.org/10.3390/app11125426