
The Role of HNO2 in the Generation of Plasma-Activated Water by Air Transient Spark Discharge



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reactor Details
2.2. TS Generation and Diagnostics
2.3. Gas Phase Diagnostics
2.4. Analysis of Water
3. Results and Discussion
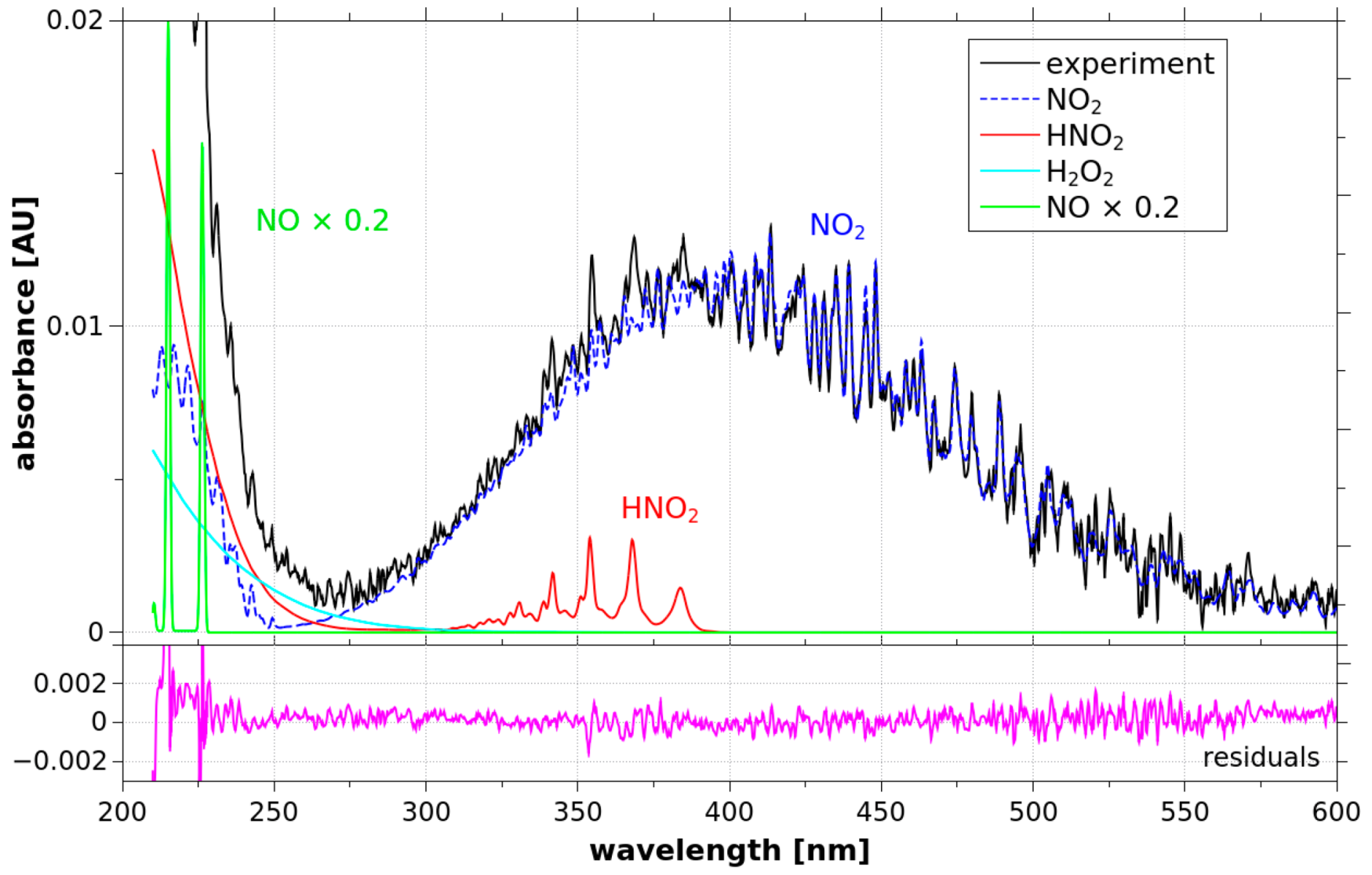
3.1. Generation of RONS in the Gas Phase
3.2. Solvation of Gaseous RONS to ES Microdroplets
3.3. Analysis of Plasma-Activated Water
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brandenburg, R.; Bogaerts, A.; Bongers, W.; Fridman, A.; Fridman, G.; Locke, B.R.; Miller, V.; Reuter, S.; Schiorlin, M.; Verreycken, T.; et al. White paper on the future of plasma science in environment, for gas conversion and agriculture. Plasma Process. Polym. 2019, 16, 1700250. [Google Scholar] [CrossRef] [Green Version]
- Cvelbar, U.; Walsh, J.L.; Cernak, M.; De Vries, H.W.; Reuter, S.; Belmonte, T.; Corbella, C.; Miron, C.; Hojnik, N.; Jurov, A.; et al. White paper on the future of plasma science and technology in plastics and textiles. Plasma Process. Polym. 2019, 16, 1700228. [Google Scholar] [CrossRef] [Green Version]
- Bekeschus, S.; Favia, P.; Robert, E.; Von Woedtke, T. White paper on plasma for medicine and hygiene: Future in plasma health sciences. Plasma Process. Polym. 2019, 16, 1800033. [Google Scholar] [CrossRef] [Green Version]
- Von Woedtke, T.; Reuter, S.; Masur, K.; Weltmann, K.-D. Plasmas for Medicine. Phys. Rep. 2013, 530, 291–320. [Google Scholar] [CrossRef]
- Keidar, M. Plasma for cancer treatment. Plasma Sources Sci. Technol. 2015, 24, 033001. [Google Scholar] [CrossRef]
- Lu, X.; Ye, T.; Cao, Y.; Sun, Z.; Xiong, Q.; Tang, Z.; Xiong, Z.; Hu, J.; Jiang, Z.; Pan, Y. The roles of the various plasma agents in the inactivation of bacteria. J. Appl. Phys. 2008, 104, 053309. [Google Scholar] [CrossRef]
- Lukes, P.; Clupek, M.; Babicky, V.; Sunka, P. Ultraviolet Radiation from the Pulsed Corona Discharge in Water. Plasma Sources Sci. Technol. 2008, 17, 024012. [Google Scholar] [CrossRef]
- Dobrynin, D.; Fridman, G.; Friedman, G.; Fridman, A. Physical and Biological Mechanisms of Direct Plasma Interaction with Living Tissue. New J. Phys. 2009, 11, 115020. [Google Scholar] [CrossRef]
- Machala, Z.; Chládeková, L.; Pelach, M. Plasma Agents in Bio-Decontamination by Dc Discharges in Atmospheric Air. J. Phys. D Appl. Phys. 2010, 43, 222001. [Google Scholar] [CrossRef]
- Janda, M.; Martišovitš, V.; Hensel, K.; Machala, Z. Generation of Antimicrobial Nox by Atmospheric Air Transient Spark Discharge. Plasma Chem. Plasma Process. 2016, 36, 767–781. [Google Scholar] [CrossRef]
- Bruggeman, P.J.; Kushner, M.J.; Locke, B.R.; Gardeniers, J.G.E.; Graham, W.G.; Graves, D.B.; Hofman-Caris, R.C.H.M.; Maric, D.; Reid, J.P.; Ceriani, E.; et al. Plasmaliquid Interactions: A Review and Roadmap. Plasma Sources Sci. Technol. 2016, 25, 053002. [Google Scholar] [CrossRef]
- Puač, N.; Gherardi, M.; Shiratani, M. Plasma Agriculture: A Rapidly Emerging Field. Plasma Process. Polym. 2018, 15, 1700174. [Google Scholar] [CrossRef]
- Thirumdas, R.; Kothakota, A.; Annapure, U.; Siliveru, K.; Blundell, R.; Gatt, R.; Valdramidis, V.P. Plasma Activated Water (PAW): Chemistry, Physico-Chemical Properties, Applications in Food and Agriculture. Trends Food Sci. Technol. 2018, 77, 21–31. [Google Scholar] [CrossRef]
- Brisset, J.-L.; Pawlat, J. Chemical Effects of Air Plasma Species on Aqueous Solutes in Direct and Delayed Exposure Modes: Discharge, Post-Discharge and Plasma Activated Water. Plasma Chem. Plasma Process. 2016, 36, 355–381. [Google Scholar] [CrossRef]
- Machala, Z.; Tarabová, B.; Sersenová, D.; Janda, M.; Hensel, K. Chemical and Antibacterial Effects of Plasma Activated Water: Correlation with Gaseous and Aqueous Reactive Oxygen and Nitrogen Species, Plasma Sources and Air Flow Conditions. J. Phys. D Appl. Phys. 2018, 52, 034002. [Google Scholar] [CrossRef]
- Kaushik, N.; Ghimire, B.; Li, Y.; Adhikari, M.; Veerana, M.; Kaushik, N.; Jha, N.; Adhikari, B.; Lee, S.-J.; Masur, K.; et al. Biological and Medical Applications of Plasma-Activated Media, Water and Solutions. Biol. Chem. 2018, 400, 39–62. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Boehm, D.; Bourke, P.; Cullen, P.J. Achieving Reactive Species Specificity Within Plasma-Activated Water Through Selective Generation Using Air Spark and Glow Discharges. Plasma Process. Polym. 2017, 14, 1600207. [Google Scholar] [CrossRef] [Green Version]
- Lukes, P.; Locke, B.R.; Brisset, J.-L. Aqueous-phase chemistry of electrical discharge plasma in water and in gas–liquid environments. In Plasma Chemistry and Catalysis in Gases and Liquids; Wiley-VCH Verlag & Co. KGaA: Weinheim, Germany, 2012; pp. 243–308. ISBN 978-3-527-64952-5. [Google Scholar]
- Lukes, P.; Dolezalova, E.; Sisrova, I.; Clupek, M. Aqueous-Phase Chemistry and Bactericidal Effects from an Air Discharge Plasma in Contact with Water: Evidence for the Formation of Peroxynitrite Through a Pseudo-Second-Order Post-Discharge Reaction of H2O2 and HNO2. Plasma Sources Sci. Technol. 2014, 23, 015019. [Google Scholar] [CrossRef]
- Huang, D.M.; Chen, Z.M. Reinvestigation of the Henry’s Law Constant for Hydrogen Peroxide with Temperature and Acidity Variation. J. Environ. Sci. 2010, 22, 570–574. [Google Scholar] [CrossRef]
- Kruszelnicki, J.; Lietz, A.M.; Kushner, M.J. Atmospheric Pressure Plasma Activation of Water Droplets. J. Phys. D Appl. Phys. 2019, 52, 355207. [Google Scholar] [CrossRef] [Green Version]
- Hassan, M.E.; Janda, M.; Machala, Z. Transport of Gaseous Hydrogen Peroxide and Ozone into Bulk Water vs. Electrosprayed Aerosol. Water 2021, 13, 182. [Google Scholar] [CrossRef]
- Stratton, G.R.; Bellona, C.L.; Dai, F.; Holsen, T.M.; Thagard, S.M. Plasma-Based Water Treatment: Conception and Application of a New General Principle for Reactor Design. Chem. Eng. J. 2015, 273, 543–550. [Google Scholar] [CrossRef]
- Zeleny, J. The Electrical Discharge from Liquid Points, and a Hydrostatic Method of Measuring the Electric Intensity at Their Surfaces. Phys. Rev. 1914, 3, 69–91. [Google Scholar] [CrossRef] [Green Version]
- Zeleny, J. Instability of Electrified Liquid Surfaces. Phys. Rev. 1917, 10, 1–6. [Google Scholar] [CrossRef]
- Dwivedi, P.; Matz, L.M.; Atkinson, D.A.; Hill, H.H., Jr. Electrospray ionization-ion mobility spectrometry: A rapid analytical method for aqueous nitrate and nitrite analysis. Analyst 2004, 129, 139–144. [Google Scholar] [CrossRef]
- Jaworek, A. Electrospray droplet sources for thin film deposition. J. Mater. Sci. 2007, 42, 266–297. [Google Scholar] [CrossRef]
- Carotenuto, C.; Di Natale, F.; Lancia, A. Wet Electrostatic Scrubbers for the Abatement of Submicronic Particulate. Chem. Eng. J. 2010, 165, 35–45. [Google Scholar] [CrossRef]
- Cui, H.; Li, N.; Peng, J.; Cheng, J.; Zhang, N.; Wu, Z. Modeling the Particle Scavenging and Thermal Efficiencies of a Heat Absorbing Scrubber. Build. Environ. 2017, 111, 218–227. [Google Scholar] [CrossRef]
- Jaworek, A.; Sobczyk, A.T.; Krupa, A. Electrospray Application to Powder Production and Surface Coating. J. Aerosol Sci. 2018, 125, 57–92. [Google Scholar] [CrossRef]
- Boda, S.K.; Li, X.; Xie, J. Electrospraying an Enabling Technology for Pharmaceutical and Biomedical Applications: A Review. J. Aerosol Sci. 2018, 125, 164–181. [Google Scholar] [CrossRef]
- Ball, A.K.; Roy, S.S.; Kisku, D.R.; Murmu, N.C.; Dos Santos Coelho, L. Optimization of Drop Ejection Frequency in EHD Inkjet Printing System Using an Improved Firefly Algorithm. Appl. Soft Comput. 2020, 94, 106438. [Google Scholar] [CrossRef]
- Kim, S.; Jung, M.; Choi, S.; Lee, J.; Lim, J.; Kim, M. Discharge Current of Water Electrospray with Electrical Conductivity under High-Voltage and High-Flow-Rate Conditions. Exp. Therm. Fluid Sci. 2020, 118, 110151. [Google Scholar] [CrossRef]
- Kim, Y.; Jung, S.; Kim, S.; Choi, S.T.; Kim, M.; Lee, H. Heat Transfer Performance of Water-Based Electrospray Cooling. Int. Commun. Heat Mass Transf. 2020, 118, 104861. [Google Scholar] [CrossRef]
- Borra, J.-P. Review on Water Electro-Sprays and Applications of Charged Drops with Focus on the Corona-Assisted Cone-Jet Mode for High Efficiency Air Filtration by Wet Electro-Scrubbing of Aerosols. J. Aerosol Sci. 2018, 125, 208–236. [Google Scholar] [CrossRef]
- Rosell-Llompart, J.; Grifoll, J.; Loscertales, I.G. Electrosprays in the Cone-Jet Mode: From Taylor Cone Formation to Spray Development. J. Aerosol Sci. 2018, 125, 2–31. [Google Scholar] [CrossRef]
- Jaworek, A.; Gañán-Calvo, A.M.; Machala, Z. Low Temperature Plasmas and Electrosprays. J. Phys. D Appl. Phys. 2019, 52, 233001. [Google Scholar] [CrossRef]
- Kanev, I.L.; Mikheev, A.Y.; Shlyapnikov, Y.M.; Shlyapnikova, E.A.; Morozova, T.Y.; Morozov, V.N. Are Reactive Oxygen Species Generated in Electrospray at Low Currents? Anal. Chem. 2014, 86, 1511–1517. [Google Scholar] [CrossRef] [PubMed]
- Burlica, R.; Grim, R.G.; Shih, K.-Y.; Balkwill, D.; Locke, B.R. Bacteria Inactivation Using Low Power Pulsed Gliding Arc Discharges with Water Spray. Plasma Process. Polym. 2010, 7, 640–649. [Google Scholar] [CrossRef]
- Oinuma, G.; Nayak, G.; Du, Y.; Bruggeman, P.J. Controlled Plasmadroplet Interactions: A Quantitative Study of OH Transfer in Plasmaliquid Interaction. Plasma Sources Sci. Technol. 2020, 29, 095002. [Google Scholar] [CrossRef]
- Kovalova, Z.; Leroy, M.; Kirkpatrick, M.J.; Odic, E.; Machala, Z. Corona Discharges with Water Electrospray for Escherichia coli Biofilm Eradication on a Surface. Bioelectrochemistry 2016, 112, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Becker, K.; Kleffmann, J.; Kurtenbach, A.; Wiesen, P. Solubility of Nitrous Acid (HONO) in Sulfuric Acid Solutions. J. Phys. Chem. 1996, 100, 14984–14990. [Google Scholar] [CrossRef]
- Lee, Y.N.; Schwartz, S.E. Reaction Kinetics of Nitrogen Dioxide with Liquid Water at Low Partial Pressure. J. Phys. Chem. 1981, 85, 840–848. [Google Scholar] [CrossRef]
- Armor, J.N. Influence of pH and Ionic Strength Upon Solubility of Nitric Oxide in Aqueous Solution. J. Chem. Eng. Data 1974, 19, 82–84. [Google Scholar] [CrossRef]
- Kučerová, K.; Machala, Z.; Hensel, K. Transient Spark Discharge Generated in Various N2/O2 Gas Mixtures: Reactive Species in the Gas and Water and Their Antibacterial Effects. Plasma Chem. Plasma Process. 2020, 40, 749–773. [Google Scholar] [CrossRef]
- Janda, M.; Martišovitš, V.; Machala, Z. Transient Spark: A Dc-Driven Repetitively Pulsed Discharge and Its Control by Electric Circuit Parameters. Plasma Sources Sci. Technol. 2011, 20, 035015. [Google Scholar] [CrossRef]
- Janda, M.; Machala, Z.; Niklová, A.; Martišovitš, V. The Streamer-to-Spark Transition in a Transient Spark: A Dc-Driven Nanosecond-Pulsed Discharge in Atmospheric Air. Plasma Sources Sci. Technol. 2012, 21, 045006. [Google Scholar] [CrossRef] [Green Version]
- Gordon, I.E.; Rothman, L.S.; Hill, C.; Kochanov, R.V.; Tan, Y.; Bernath, P.F.; Birk, M.; Boudon, V.; Campargue, A.; Chance, K.V.; et al. The HITRAN2016 Molecular Spectroscopic Database. J. Quant. Spectrosc. Radiat. Transf. 2017, 203, 3–69. [Google Scholar] [CrossRef]
- Kochanov, R.V.; Gordon, I.E.; Rothman, L.S.; Shine, K.P.; Sharpe, S.W.; Johnson, T.J.; Wallington, T.J.; Harrison, J.J.; Bernath, P.F.; Birk, M.; et al. Infrared Absorption Cross-Sections in HITRAN2016 and Beyond: Expansion for Climate, Environment, and Atmospheric Applications. J. Quant. Spectrosc. Radiat. Transf. 2019, 230, 172–221. [Google Scholar] [CrossRef]
- Keller-Rudek, H.; Moortgat, G.K.; Sander, R.; Sörensen, R. The MPI-Mainz UV/VIS Spectral Atlas of Gaseous Molecules of Atmospheric Interest. Earth Syst. Sci. Data 2013, 5, 365–373. [Google Scholar] [CrossRef] [Green Version]
- Oehmigen, K.; Hähnel, M.; Brandenburg, R.; Wilke, C.; Weltmann, K.-D.; Von Woedtke, T. The Role of Acidification for Antimicrobial Activity of Atmospheric Pressure Plasma in Liquids. Plasma Process. Polym. 2010, 7, 250–257. [Google Scholar] [CrossRef]
- Chauvin, J.; Judée, F.; Yousfi, M.; Vicendo, P.; Merbahi, N. Analysis of Reactive Oxygen and Nitrogen Species Generated in Three Liquid Media by Low Temperature Helium Plasma Jet. Sci. Rep. 2017, 7, 4562. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.E.; Cha, N.R.; Lindsay, A.D.; Clark, D.S.; Graves, D.B. The Role of Interfacial Reactions in Determining Plasma–Liquid Chemistry. Plasma Chem. Plasma Process. 2016, 36, 1393–1415. [Google Scholar] [CrossRef]
- Wende, K.; Williams, P.; Dalluge, J.; Gaens, W.V.; Aboubakr, H.; Bischof, J.; Von Woedtke, T.; Goyal, S.M.; Weltmann, K.D.; Bogaerts, A.; et al. Identification of the biologically active liquid chemistry induced by a nonthermal atmospheric pressure plasma jet. Biointerphases 2015, 10, 029518. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Ma, R.; Zhang, Q.; Feng, H.; Liang, Y.; Zhang, J.; Fang, J. Assessment of the Physicochemical Properties and Biological Effects of Water Activated by Non-Thermal Plasma above and Beneath the Water Surface. Plasma Process. Polym. 2015, 12, 439–449. [Google Scholar] [CrossRef]
- Lu, P.; Boehm, D.; Cullen, P.; Bourke, P. Controlled Cytotoxicity of Plasma Treated Water Formulated by Open-Air Hybrid Mode Discharge. Appl. Phys. Lett. 2017, 110, 264102. [Google Scholar] [CrossRef] [Green Version]
- Kovačević, V.V.; Dojčinović, B.P.; Jović, M.; Roglić, G.M.; Obradović, B.M.; Kuraica, M.M. Measurement of Reactive Species Generated by Dielectric Barrier Discharge in Direct Contact with Water in Different Atmospheres. J. Phys. D Appl. Phys. 2017, 50, 155205. [Google Scholar] [CrossRef]
- Tarabová, B.; Lukeš, P.; Janda, M.; Hensel, K.; Šikurová, L.; Machala, Z. Specificity of Detection Methods of Nitrites and Ozone in Aqueous Solutions Activated by Air Plasma. Plasma Process. Polym. 2018, 15, 1800030. [Google Scholar] [CrossRef]
- Eisenberg, G. Colorimetric Determination of Hydrogen Peroxide. Ind. Eng. Chem. Anal. Ed. 1943, 15, 327–328. [Google Scholar] [CrossRef]
- Janda, M.; Machala, Z.; Lacoste, D.; Stancu, G.D.; Laux, C.O. Stabilization of A Lean Methane-Air Flame Using Transient Spark Discharge. In Proceedings of the 13th International Symposium on High Pressure Low Temperature Plasma Chemistry (Hakone XIII), Kazimierz Dolny, Poland, 9–14 September 2012; pp. 185–189. [Google Scholar]
- Borra, J.-P.; Ehouarn, P.; Boulaud, D. Electrohydrodynamic Atomisation of Water Stabilised by Glow Discharge—Operating Range and Droplet Properties. J. Aerosol Sci. 2004, 35, 1313–1332. [Google Scholar] [CrossRef]
- Janda, M.; Hensel, K.; Machala, Z. Chemical Kinetic Model of Transient Spark: Spark Phase and NOx Formation. In Proceedings of the 22nd Symposium on Applications of Plasma Processes and 11th Eu-Japan Joint Symposium on Plasma Processing Sapp XXII, Strbske Pleso, Slovakia, 18–24 January 2019; pp. 25–32. [Google Scholar]
- Capitelli, M.; Ferreira, C.M.; Gordiets, B.F.; Osipov, A.I. Plasma Kinetics in Atmospheric Gases; Springer: Berlin/Heidelberg, Germany, 2000. [Google Scholar]
- Janda, M.; Martišovitš, V.; Hensel, K.; Dvonč, L.; Machala, Z. Measurement of the Electron Density in Transient Spark Discharge. Plasma Sources Sci. Technol. 2014, 23, 065016. [Google Scholar] [CrossRef] [Green Version]
- Herron, J. Evaluated Chemical Kinetics Data for Reactions of N(2D), N(2P), and N2(A3∑u+) in the Gas Phase. J. Phys. Chem. Ref. Data 1999, 28, 1453–1483. [Google Scholar] [CrossRef]
- Melton, C.E. Cross Sections and Interpretation of Dissociative Attachment Reactions Producing OH−, O−, and H− in H2O. J. Chem. Phys. 1972, 57, 4218–4225. [Google Scholar] [CrossRef]
- Lowke, J.J.; Morrow, R. Theoretical analysis of removal of oxides of sulphur and nitrogen in pulsed operation of electrostatic precipitators. IEEE Trans. Plasma Sci. 1995, 23, 661–671. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janda, M.; Hensel, K.; Tóth, P.; Hassan, M.E.; Machala, Z. The Role of HNO2 in the Generation of Plasma-Activated Water by Air Transient Spark Discharge. Appl. Sci. 2021, 11, 7053. https://doi.org/10.3390/app11157053
Janda M, Hensel K, Tóth P, Hassan ME, Machala Z. The Role of HNO2 in the Generation of Plasma-Activated Water by Air Transient Spark Discharge. Applied Sciences. 2021; 11(15):7053. https://doi.org/10.3390/app11157053
Chicago/Turabian StyleJanda, Mário, Karol Hensel, Peter Tóth, Mostafa E. Hassan, and Zdenko Machala. 2021. "The Role of HNO2 in the Generation of Plasma-Activated Water by Air Transient Spark Discharge" Applied Sciences 11, no. 15: 7053. https://doi.org/10.3390/app11157053