
Contribution to the Understanding of Protein–Protein Interface and Ligand Binding Site Based on Hydrophobicity Distribution—Application to Ferredoxin I and II Cases



Abstract
:Featured Application
Abstract
1. Introduction
2. Materials and Methods
2.1. Data
2.2. FOD Model
- O—observed hydrophobicity distribution based on pairwise hydrophobic interactions between residues, calculated using Michael Levitt’s polynomial [34];
- T—theoretical hydrophobicity distribution calculated using 3D Gauss capsule fit to the molecule (Figure S1), based on location of residues in respect to this “drop”.
2.3. MIR Model
2.4. Tools and Websites
3. Results
3.1. Sequence Analysis
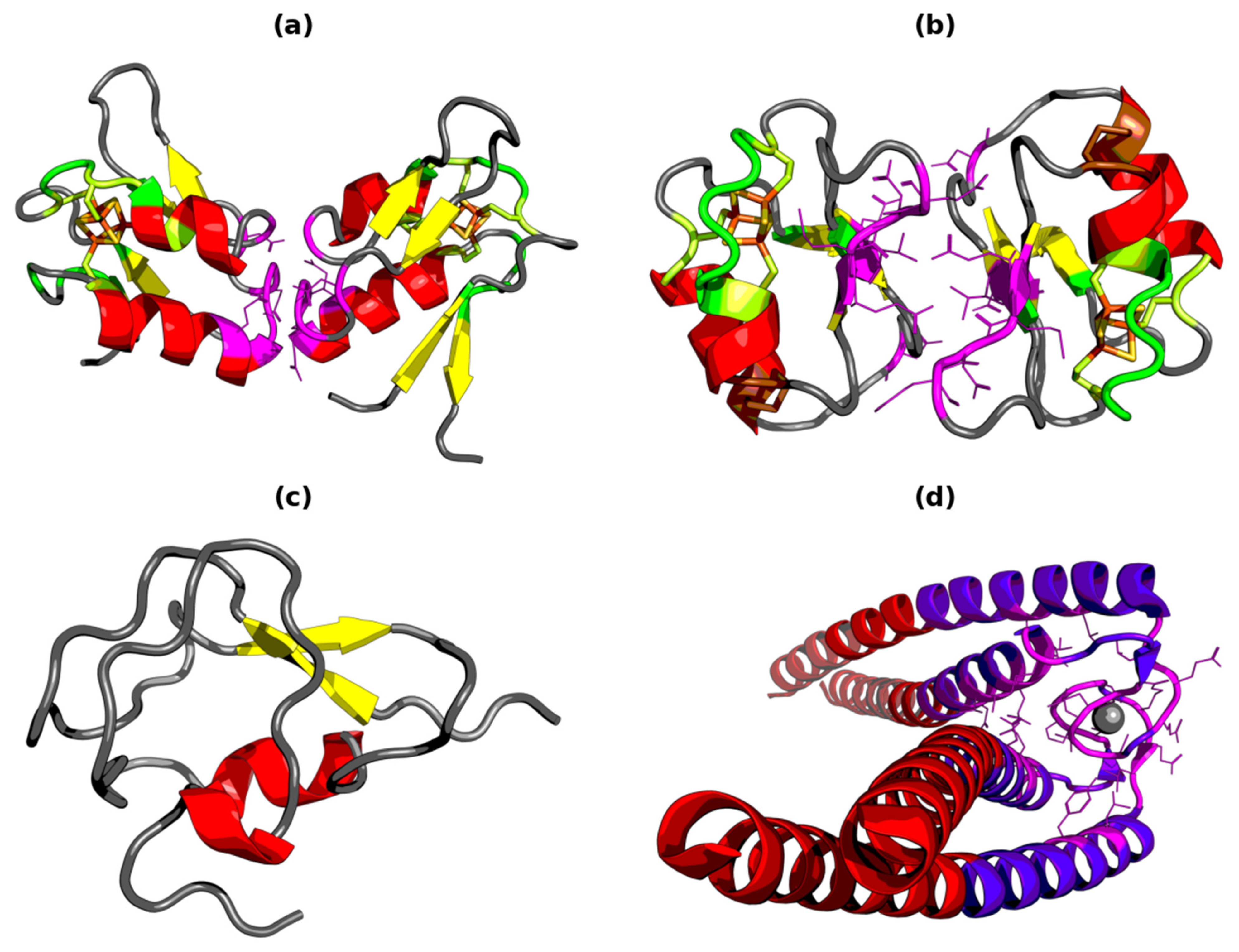
3.2. Structure Analysis
3.3. Hydrophobicity Characteristic—Monomers
3.4. Hydrophobicity Characteristic—Dimers
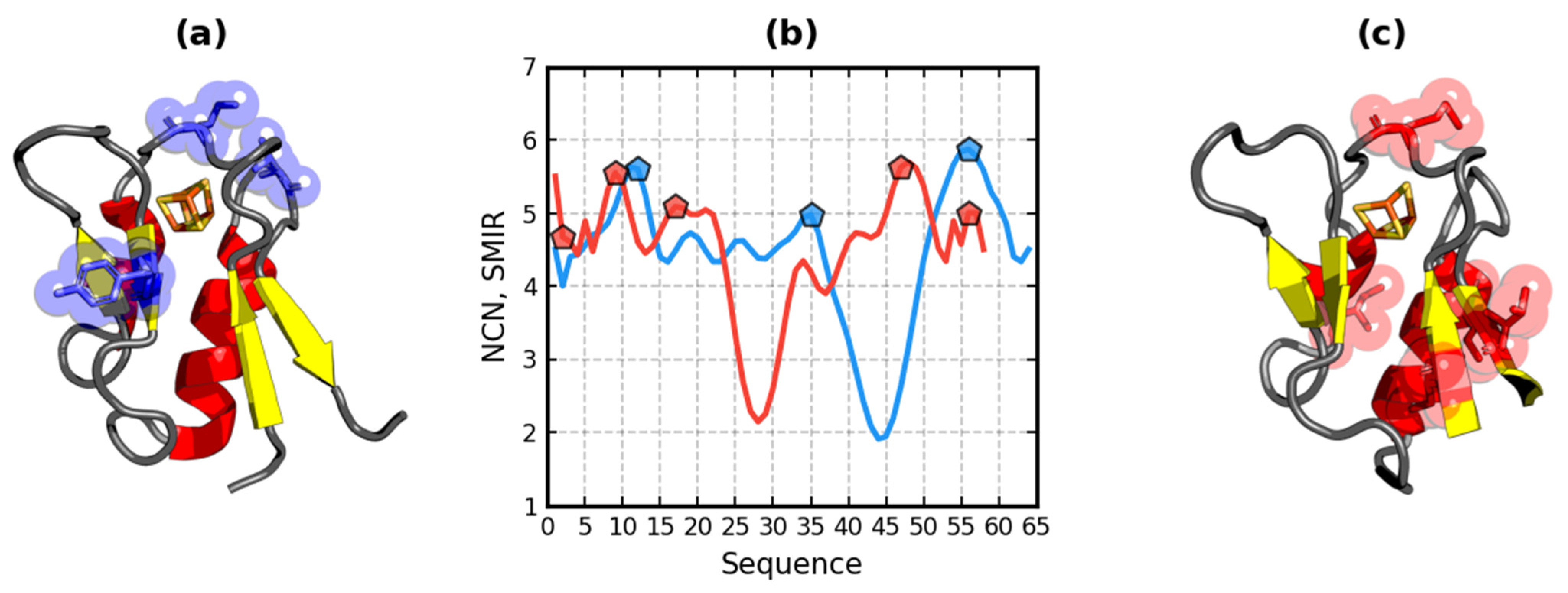
3.5. SMIR Analysis
3.6. Hydrophobicity-Based Ligand Binding Site and Protein–Protein Interface Determination
- Class C (core)—hydrophobic core: both T and O are relatively high (T↑↑O) and the difference between them is relatively low (|T-O|→0);
- Class S (surf)—hydrophilic surface: both T and O are relatively low (T↓↓O) and the difference between them is relatively low (|T-O|→0);
- Class B (bind)—deficiency of hydrophobicity closer to the center of the molecule, hinting a possible ligand binding pocket: T is relatively high, O is relatively low (T↑↓O) and the difference between them is relatively high (|T-O|→1);
- Class D (dock)—excess of hydrophobicity closer to the outside of the molecule, hinting a possible protein docking interface: T is relatively low, O is relatively high (T↓↑O) and the difference between them is relatively high (|T-O|→1);
- Class Z (zero)—neither of the above: low difference between T and O (T ≈ O) but also unremarkable position on the T vs. O map (near the average, somewhere in between core and surface)—a model-accordant but hydrophobically insignificant residue.
- Level 3 (classes C3, S3, B3 and D3)—most prominent class members, with strongest defining features, i.e., lowest |T-O| for C and S and highest |T-O| for B and D;
- Level 2 (classes C2, S2, B2 and D2)—significant class members but not as outstanding on the map as level 3, i.e., low |T-O| for C and S and high |T-O| for B and D;
- Level 1 (classes C1, S1, B1 and D1)—weak class members, extracted from class Z.
- (Figure 7a) plot residues as points in T vs. O space (T on the X-axis, O on the Y-axis); calculate quartiles of the T distribution: Q2 (median), Q1 (median of T < Q2) and Q3 (median of T > Q2); assign three thresholds: T1 = Q1, T3 = Q3, T2 = (Q1 + Q3)/2;
- (Figure 7b) draw a T = O line and shift it to point [T1,T3]; draw it again and shift it to point [T3,T1]; draw a T = -O line and shift it to point [T1,T1]; draw it again and shift it to point [T3,T3]; four points where pairs of these lines intersect are the class Z square vertexes: vcd, vds, vsb and vbc (meaning of indexes is explained in point 5);
- (Figure 7c) extend lines away from class Z square vertexes, partitioning the T vs. O space into five segments, with class Z square in the middle of the map;
- (Figure 7d) place four helper circles symmetrically around class Z square to cut away portions from it to be used as delimiters for level 1 class zones; each circle has T3-T1 radius and intersects two nearby class Z square vertexes (i.e., vcd and vds);
- (Figure 7e) remove helper circles except for their arcs within class Z square; give space segments around it following labels: C (core, top-right), D (dock, top-left), S (surf, bottom-left) and B (bind, bottom-right); indexes at names of class Z square vertexes inform between which classes they are located, i.e., vcd is between C and D;
- (Figure 7f) draw separation lines (vds to vcd and vsb to vbc) to demarcate levels within each class; in case of C and S, level 3 is closer (via orthogonal projection) to T = O line than 50% of length of edge of class Z square (i.e., half of the distance between vcd and vbc, shown as dashed lines) and level 2 is outside this range; in case of B and D, level 2 is also outside this range, while level 3 is even further away from T = O line: more than 75% of length of edge of class Z square (shown as dotted lines).
3.7. Ligand Binding Site and Protein–Protein Interface Determination in Ferredoxin I and II
3.8. Comparative Analysis—Type III Antifreeze Protein and Rad50 Domain of Mre11
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Burkhart, B.W.; Febvre, H.P.; Santangelo, T.J. Distinct Physiological Roles of the Three Ferredoxins Encoded in the Hyperthermophilic Archaeon Thermococcus kodakarensis. mBio 2019, 10, e02807-18. [Google Scholar] [CrossRef] [Green Version]
- Maiocco, S.J.; Arcinas, A.J.; Booker, S.J.; Elliott, S.J. Parsing redox potentials of five ferredoxins found within Thermotoga maritima. Protein Sci. 2018, 28, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Grinberg, A.V.; Hannemann, F.; Schiffler, B.; Müller, J.; Heinemann, U.; Bernhardt, R. Adrenodoxin: Structure, stability, and electron transfer properties. Proteins Struct. Funct. Bioinform. 2000, 40, 590–612. [Google Scholar] [CrossRef]
- Hsieh, Y.-C.; Liu, M.-Y.; Le Gall, J.; Chen, C.-J. Anaerobic purification and crystallization to improve the crystal quality: Ferredoxin II from Desulfovibrio gigas. Acta Crystallogr. Sect. D Biol. Crystallogr. 2005, 61, 780–783. [Google Scholar] [CrossRef]
- Im, S.; Liu, G.; Luchinat, C.; Sykes, A.G.; Bertini, I. The solution structure of parsley [2Fe-2S]ferredoxin. Eur. J. Biochem. 1998, 258, 465–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raanan, H.; Pike, D.H.; Moore, E.K.; Falkowski, P.G.; Nanda, V. Modular origins of biological electron transfer chains. Proc. Natl. Acad. Sci. USA 2018, 115, 1280–1285. [Google Scholar] [CrossRef] [Green Version]
- Marco, P.; Kozuleva, M.; Eilenberg, H.; Mazor, Y.; Gimeson, P.; Kanygin, A.; Redding, K.; Weiner, I.; Yacoby, I. Binding of ferredoxin to algal photosystem I involves a single binding site and is composed of two thermodynamically distinct events. Biochim. Biophys. Acta (BBA)—Bioenerg. 2018, 1859, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Beilke, D.; Weiss, R.; Löhr, F.; Pristovšek, P.; Hannemann, F.; Bernhardt, R.; Rüterjans, H. A New Electron Transport Mechanism in Mitochondrial Steroid Hydroxylase Systems Based on Structural Changes upon the Reduction of Adrenodoxin†. Biochemistry 2002, 41, 7969–7978. [Google Scholar] [CrossRef]
- Goodfellow, B.; Macedo, A.L.; Rodrigues, P.; Moura, I.; Wray, V.; Moura, J.J.G. The solution structure of a [3Fe-4S] ferredoxin: Oxidised ferredoxin II from Desulfovibrio gigas. JBIC J. Biol. Inorg. Chem. 1999, 4, 421–430. [Google Scholar] [CrossRef]
- Konieczny, L.; Roterman, I. Globular or ribbon-like micelle. In From Globular Proteins to Amyloids; Elsevier: Amsterdam, The Netherlands, 2020; pp. 41–54. [Google Scholar] [CrossRef]
- Gadzała, M.; Dułak, D.; Kalinowska, B.; Baster, Z.; Bryliński, M.; Konieczny, L.; Banach, M.; Roterman, I. The aqueous environment as an active participant in the protein folding process. J. Mol. Graph. Model. 2019, 87, 227–239. [Google Scholar] [CrossRef]
- Banach, M.; Konieczny, L.; Roterman, I. Why do antifreeze proteins require a solenoid? Biochimie 2018, 144, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Kalinowska, B.; Banach, M.; Wiśniowski, Z.; Konieczny, L.; Roterman, I. Is the hydrophobic core a universal structural element in proteins? J. Mol. Model. 2017, 23, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banach, M.; Konieczny, L.; Roterman, I. The fuzzy oil drop model, based on hydrophobicity density distribution, generalizes the influence of water environment on protein structure and function. J. Theor. Biol. 2014, 359, 6–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roterman, I.; Banach, M.; Konieczny, L. Application of the Fuzzy Oil Drop Model Describes Amyloid as a Ribbonlike Micelle. Entropy 2017, 19, 167. [Google Scholar] [CrossRef] [Green Version]
- Banach, M.; Konieczny, L.; Roterman, I. The Amyloid as a Ribbon-Like Micelle in Contrast to Spherical Micelles Represented by Globular Proteins. Molecules 2019, 24, 4395. [Google Scholar] [CrossRef] [Green Version]
- Andrusier, N.; Mashiach, E.; Nussinov, R.; Wolfson, H.J. Principles of flexible protein-protein docking. Proteins Struct. Funct. Bioinform. 2008, 73, 271–289. [Google Scholar] [CrossRef] [Green Version]
- Janin, J. Welcome to CAPRI: A Critical Assessment of PRedicted Interactions. Proteins Struct. Funct. Bioinform. 2002, 47, 257. [Google Scholar] [CrossRef]
- Available online: https://www.ebi.ac.uk/pdbe/complex-pred/capri (accessed on 23 June 2021).
- Berman, H.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.rcsb.org (accessed on 18 October 2020).
- Sillitoe, I.; Dawson, N.; Lewis, T.E.; Das, S.; Lees, J.G.; Ashford, P.; Tolulope, A.; Scholes, H.; Senatorov, I.; Bujan, A.; et al. CATH: Expanding the horizons of structure-based functional annotations for genome sequences. Nucleic Acids Res. 2019, 47, D280–D284. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.cathdb.info (accessed on 22 October 2020).
- Sery, A.; Housset, D.; Serre, L.; Bonicel, J.; Hatchikian, C.; Frey, M.; Roth, M. Crystal Structure of the Ferredoxin I from Desulfovibrio africanus at 2.3-.ANG. Resolution. Biochemistry 1994, 33, 15408–15417. [Google Scholar] [CrossRef]
- Kissinger, C.R.; Sieker, L.C.; Adman, E.T.; Jensen, L.H. Refined crystal structure of ferredoxin II from Desulfovibrio gigas at 1.7 Å. J. Mol. Biol. 1991, 219, 693–715. [Google Scholar] [CrossRef]
- Graether, S.P.; DeLuca, C.I.; Baardsnes, J.; Hill, G.A.; Davies, P.L.; Jia, Z. Quantitative and Qualitative Analysis of Type III Antifreeze Protein Structure and Function. J. Biol. Chem. 1999, 274, 11842–11847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopfner, K.-P.; Craig, L.; Moncalian, G.; Zinkel, R.A.; Usui, T.; Owen, B.A.L.; Karcher, A.; Henderson, B.; Bodmer, J.-L.; McMurray, C.T.; et al. The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature 2002, 418, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Available online: https://www.ebi.ac.uk/pdbsum (accessed on 19 March 2021).
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2020, 49, D412–D419. [Google Scholar] [CrossRef]
- Kalinowska, B.; Banach, M.; Konieczny, L.; Roterman, I. Application of Divergence Entropy to Characterize the Structure of the Hydrophobic Core in DNA Interacting Proteins. Entropy 2015, 17, 1477–1507. [Google Scholar] [CrossRef] [Green Version]
- Banach, M.; Fabian, P.; Stapor, K.; Konieczny, L.; Roterman, A.I. Structure of the Hydrophobic Core Determines the 3D Protein Structure—Verification by Single Mutation Proteins. Biomolecules 2020, 10, 767. [Google Scholar] [CrossRef]
- Konieczny, L.; Roterman, I. Description of the fuzzy oil drop model. In From Globular Proteins to Amyloids; Elsevier: Amsterdam, The Netherlands, 2020; pp. 1–11. [Google Scholar] [CrossRef]
- Levitt, M. A simplified representation of protein conformations for rapid simulation of protein folding. J. Mol. Biol. 1976, 104, 59–107. [Google Scholar] [CrossRef]
- Banach, M.; Konieczny, L.; Roterman, I. The active site in a single-chain enzyme. In From Globular Proteins to Amyloids; Elsevier: Amsterdam, The Netherlands, 2020; pp. 71–78. [Google Scholar] [CrossRef]
- Kullback, S.; Leibler, R.A. On Information and Sufficiency. Ann. Math. Stat. 1951, 22, 79–86. [Google Scholar] [CrossRef]
- Banach, M.; Konieczny, L.; Roterman, I. Composite structures. In From Globular Proteins to Amyloids; Elsevier: Amsterdam, The Netherlands, 2020; pp. 117–133. [Google Scholar] [CrossRef]
- Fabian, P.; Banach, M.; Stapor, K.; Konieczny, L.; Ptak-Kaczor, M.; Roterman, I. The Structure of Amyloid versus the Structure of Globular Proteins. Int. J. Mol. Sci. 2020, 21, 4683. [Google Scholar] [CrossRef]
- Dułak, D.; Gadzała, M.; Stapor, K.; Fabian, P.; Konieczny, L.; Roterman, I. Folding with active participation of water. In From Globular Proteins to Amyloids; Elsevier: Amsterdam, The Netherlands, 2020; pp. 13–26. [Google Scholar] [CrossRef]
- Banach, M.; Konieczny, L.; Roterman, I. Protein-protein interaction encoded as an exposure of hydrophobic residues on the surface. In From Globular Proteins to Amyloids; Elsevier: Amsterdam, The Netherlands, 2020; pp. 79–89. [Google Scholar] [CrossRef]
- Banach, M.; Konieczny, L.; Roterman, I. Ligand binding cavity encoded as a local hydrophobicity deficiency. In From Globular Proteins to Amyloids; Elsevier: Amsterdam, The Netherlands, 2020; pp. 91–93. [Google Scholar] [CrossRef]
- Papandreou, N.; Berezovsky, I.N.; Lopes, A.; Eliopoulos, E.; Chomilier, J. Universal positions in globular proteins: From observation to simulation. Eur. J. Biochem. 2004, 271, 4762–4768. [Google Scholar] [CrossRef]
- Prudhomme, N.; Chomilier, J. Prediction of the protein folding core: Application to the immunoglobulin fold. Biochimie 2009, 91, 1465–1474. [Google Scholar] [CrossRef] [PubMed]
- Banach, M.; Prudhomme, N.; Carpentier, M.; Duprat, E.; Papandreou, N.; Kalinowska, B.; Chomilier, J.; Roterman, I. Contribution to the Prediction of the Fold Code: Application to Immunoglobulin and Flavodoxin Cases. PLoS ONE 2015, 10, e0125098. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System; Version 2.0; Schrödinger, LLC: New York, NY, USA.
- Available online: https://pymol.org (accessed on 5 October 2020).
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, T. MATRAS: A program for protein 3D structure comparison. Nucleic Acids Res. 2003, 31, 3367–3369. [Google Scholar] [CrossRef] [Green Version]
- Available online: http://strcomp.protein.osaka-u.ac.jp/matras (accessed on 28 October 2020).
- Sanner, M.F.; Olson, A.J.; Spehner, J.-C. Reduced surface: An efficient way to compute molecular surfaces. Biopolymers 1996, 38, 305–320. [Google Scholar] [CrossRef]
- Available online: http://mgltools.scripps.edu/packages/MSMS (accessed on 25 June 2021).
- Shanthirabalan, S.; Chomilier, J.; Carpentier, M. Structural effects of point mutations in proteins. Proteins Struct. Funct. Bioinform. 2018, 86, 853–867. [Google Scholar] [CrossRef] [Green Version]
- Carpentier, M.; Chomilier, J. Protein multiple alignments: Sequence-based versus structure-based programs. Bioinformatics 2019, 35, 3970–3980. [Google Scholar] [CrossRef]
- Konieczny, L.; Roterman, I. Information encoded in protein structure. In From Globular Proteins to Amyloids; Elsevier: Amsterdam, The Netherlands, 2020; pp. 27–39. [Google Scholar] [CrossRef]
- Shindyalov, I.N.; Bourne, P.E. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. Des. Sel. 1998, 11, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Dygut, J.; Kalinowska, B.; Banach, M.; Piwowar, M.; Konieczny, L.; Roterman, I. Structural Interface Forms and Their Involvement in Stabilization of Multidomain Proteins or Protein Complexes. Int. J. Mol. Sci. 2016, 17, 1741. [Google Scholar] [CrossRef] [Green Version]
- Banach, M.; Stapor, K.; Konieczny, L.; Fabian, P.; Roterman, I. Downhill, Ultrafast and Fast Folding Proteins Revised. Int. J. Mol. Sci. 2020, 21, 7632. [Google Scholar] [CrossRef]
- Roterman, I.; Stapor, K.; Fabian, P.; Konieczny, L.; Banach, M. Model of Environmental Membrane Field for Transmembrane Proteins. Int. J. Mol. Sci. 2021, 22, 3619. [Google Scholar] [CrossRef] [PubMed]
- Banach, M.; Kalinowska, B.; Konieczny, L.; Roterman, I. Role of Disulfide Bonds in Stabilizing the Conformation of Selected Enzymes—An Approach Based on Divergence Entropy Applied to the Structure of Hydrophobic Core in Proteins. Entropy 2016, 18, 67. [Google Scholar] [CrossRef] [Green Version]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.; Cang, Z.; Tong, Y.; Wei, G.-W. Protein pocket detection via convex hull surface evolution and associated Reeb graph. Bioinformatics 2018, 34, i830–i837. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | PDB Code | Organism | Quaternary Structure | Chain Length | Ligand | CATH Domain | Ref. |
|---|---|---|---|---|---|---|---|
| Ferredoxin I (FdI) | 1FXR | Desulfovibrio africanus | homodimer, symmetric | 64 aa | [4F4S] | 3.30.70.20 | [24] |
| Ferredoxin II (FdII) | 1FXD | Desulfovibrio gigas | 58 aa | [3F4S] | [25] | ||
| Antifreeze type III (AFP) | 9MSI | Macrozoarces americanus | monomer | 66 aa | none | 3.90.1210.10 | [26] |
| Rad50 domain (Mre11 complex) | 1L8D | Pyrococcus furiosus | homodimer, symmetric | 103 aa | Hg2+ | 1.10.287.510 | [27] |
| Fragment | Residues | RD | TvH | OvT | OvH | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| FdI | FdII | FdI | FdII | FdI | FdII | FdI | FdII | FdI | FdII | |
| monomer | chain A | chain A | 0.33 | 0.27 | 0.56 | 0.60 | 0.72 | 0.78 | 0.79 | 0.86 |
| strand A.1 | 3–6 | 1–4 | 0.18 | 0.25 | 0.91 | 0.70 | 0.98 | 0.86 | 0.86 | 0.97 |
| strand A.2 | 59–62 | 55–58 | 0.19 | 0.15 | 0.82 | 0.79 | 0.99 | 0.95 | 0.76 | 0.93 |
| sheet A | strand A.1 + A.2 | strand A.1 + A.2 | 0.21 | 0.18 | 0.83 | 0.69 | 0.94 | 0.91 | 0.77 | 0.87 |
| strand B.1 | 25–27 | 22–24 | 0.04 | 0.18 | 0.46 | 0.71 | 1.00 | 0.97 | 0.49 | 0.86 |
| strand B.2 | 34–36 | 31–33 | 0.83 | 0.89 | −0.48 | −1.00 | 0.84 | −0.99 | −0.88 | 0.99 |
| sheet B | strand B.1 + B.2 | strand B.1 + B.2 | 0.32 | 0.26 | 0.20 | 0.53 | 0.73 | 0.83 | 0.45 | 0.86 |
| all sheets | sheet A + B | sheet A + B | 0.23 | 0.19 | 0.73 | 0.64 | 0.90 | 0.90 | 0.71 | 0.84 |
| no sheets | !(sheet A + B) | !(sheet A + B) | 0.33 | 0.26 | 0.45 | 0.57 | 0.72 | 0.81 | 0.80 | 0.87 |
| helix 1 | 15–21 | 13–17 | 0.23 | 0.18 | 0.85 | 0.81 | 0.97 | 0.94 | 0.88 | 0.96 |
| helix 2 | 43–54 | 41–49 | 0.35 | 0.28 | 0.59 | 0.70 | 0.71 | 0.82 | 0.93 | 0.92 |
| all helices | helix 1 + 2 | helix 1 + 2 | 0.31 | 0.25 | 0.70 | 0.76 | 0.81 | 0.88 | 0.90 | 0.94 |
| no helices | !(helix 1 + 2) | !(helix 1 + 2) | 0.34 | 0.29 | 0.55 | 0.56 | 0.70 | 0.74 | 0.74 | 0.84 |
| P-P | FdI(P-P) | FdII(P-P) | 0.46 | 0.18 | 0.24 | 0.65 | 0.33 | 0.87 | 0.92 | 0.91 |
| no P-P | !(FdI(P-P)) | !(FdI(P-P)) | 0.31 | 0.28 | 0.56 | 0.59 | 0.75 | 0.77 | 0.79 | 0.85 |
| P-L | FdI(P-L) | FdII(P-L) | 0.40 | 0.45 | 0.37 | 0.07 | 0.67 | 0.63 | 0.84 | 0.69 |
| no P-L | !(FdI(P-L)) | !(FdII(P-L)) | 0.30 | 0.27 | 0.62 | 0.66 | 0.75 | 0.79 | 0.73 | 0.87 |
| catalytic | 11,14,17,54 | 8,14,50 | 0.25 | 0.27 | n/a | n/a | 0.90 | 0.92 | n/a | n/a |
| no catalytic | !(11,14,17,54) | !(8,14,50) | 0.33 | 0.28 | 0.58 | 0.59 | 0.72 | 0.77 | 0.76 | 0.85 |
| S-S bond | n/a | 18–42 | n/a | 0.25 | n/a | 0.62 | n/a | 0.78 | n/a | 0.90 |
| no S-S bond | n/a | !(18–42) | n/a | 0.30 | n/a | 0.58 | n/a | 0.78 | n/a | 0.83 |
| Fragment | Residues | RD | TvH | OvT | OvH | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| FdI | FdII | FdI | FdII | FdI | FdII | FdI | FdII | FdI | FdII | |
| monomer | chain A | chain A | 0.56 | 0.58 | 0.16 | 0.25 | 0.53 | 0.31 | 0.75 | 0.87 |
| strand A.1 | 3–6 | 1–4 | 0.43 | 0.32 | 0.72 | 0.56 | 0.46 | 0.75 | 0.86 | 0.96 |
| strand A.2 | 59–62 | 55–58 | 0.25 | 0.19 | 0.91 | 0.92 | 0.91 | 1.00 | 0.76 | 0.93 |
| sheet A | strand A.1 + A.2 | strand A.1 + A.2 | 0.37 | 0.38 | 0.70 | 0.35 | 0.57 | 0.58 | 0.77 | 0.87 |
| strand B.1 | 25–27 | 22–24 | 0.19 | 0.52 | 0.18 | −0.52 | 0.95 | 0.00 | 0.49 | 0.85 |
| strand B.2 | 34–36 | 31–33 | 0.97 | 0.19 | 0.97 | 0.99 | −0.73 | 0.89 | −0.88 | 0.94 |
| sheet B | strand B.1 + B.2 | strand B.1 + B.2 | 0.35 | 0.50 | 0.42 | 0.11 | 0.80 | 0.35 | 0.45 | 0.87 |
| all sheets | sheet A + B | sheet A + B | 0.33 | 0.38 | 0.55 | 0.31 | 0.71 | 0.64 | 0.71 | 0.83 |
| no sheets | !(sheet A + B) | !(sheet A + B) | 0.59 | 0.57 | 0.10 | 0.14 | 0.50 | 0.20 | 0.76 | 0.88 |
| helix 1 | 15–21 | 13–17 | 0.27 | 0.26 | 0.59 | 0.74 | 0.80 | 0.83 | 0.88 | 0.95 |
| helix 2 | 43–54 | 41–49 | 0.64 | 0.19 | −0.09 | 0.81 | 0.01 | 0.86 | 0.92 | 0.92 |
| all helices | helix 1 + 2 | helix 1 + 2 | 0.54 | 0.19 | 0.20 | 0.78 | 0.35 | 0.86 | 0.90 | 0.94 |
| no helices | !(helix 1 + 2) | !(helix 1 + 2) | 0.57 | 0.55 | 0.16 | 0.24 | 0.59 | 0.39 | 0.69 | 0.85 |
| P-P | FdI(P-P) | FdII(P-P) | 0.29 | 0.28 | 0.90 | 0.50 | 0.95 | 0.82 | 0.97 | 0.89 |
| no P-P | !(FdI(P-P)) | !(FdI(P-P)) | 0.54 | 0.57 | 0.27 | 0.30 | 0.51 | 0.34 | 0.78 | 0.86 |
| P-L | FdI(P-L) | FdII(P-L) | 0.77 | 0.65 | −0.28 | −0.03 | 0.06 | 0.30 | 0.81 | 0.69 |
| no P-L | !(FdI(P-L)) | !(FdII(P-L)) | 0.41 | 0.57 | 0.35 | 0.34 | 0.73 | 0.36 | 0.68 | 0.88 |
| catalytic | 11,14,17,54 | 8,14,50 | 0.76 | 0.68 | n/a | n/a | 0.84 | 0.38 | n/a | n/a |
| no catalytic | !(11,14,17,54) | !(8,14,50) | 0.52 | 0.58 | 0.23 | 0.27 | 0.60 | 0.35 | 0.72 | 0.86 |
| S-S bond | n/a | 18–42 | n/a | 0.51 | n/a | 0.33 | n/a | 0.42 | n/a | 0.91 |
| no S-S bond | n/a | !(18–42) | n/a | 0.53 | n/a | 0.35 | n/a | 0.34 | n/a | 0.83 |
| Class | Cluster | NativeP-PContacts | NativeP-LContacts |
| 23, 38, 40, 41, 42, 43, 46 | 6, 11, 12, 13, 14, 15, 17, 54, 56, 58, 59 | ||
| D2 | 10, 54, 55, 56, 57, 58 | ∅ | (11), 54, 56, 58, (59) |
| 21, 22, 23, 24, 25, 37, 38, 39, 40, 41, 42 | 23, 38, 40, 41, 42, (43) | ∅ | |
| D2 + D1 | 10, 11, 12, 13, 14, 15, 16, 54, 55, 56, 57, 58 | ∅ | 11, 12, 13, 14, 15, (17), 54, 56, 58, (59) |
| 21, 22, 23, 24, 25, 37, 38, 39, 40, 41, 42, 43, 46 | 23, 38, 40, 41, 42, 43, 46 | ∅ | |
| B2 + B1 | 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 20, 21, 33, 35, 36, 39, 42, 43, 44, 45, 46, 48, 49, 50, 51, 52, 53, 60, 61 | (38), (40), (41), 42, 43, 46 | 6, 11, (12), (54), (59) |
| Class | Cluster | NativeP-PContacts | NativeP-LContacts |
| 3, 23, 24, 25, 26, 32, 37 | 8, 9, 10, 11, 12, 13, 14, 31, 55, 50 | ||
| D2 + D1 | 7, 8, 9, 10, 11, 50, 51, 52, 53, 54 | ∅ | 8, 9, 10, 11, (12), 50, (55) |
| B2 | 4, 5, 6, 8, 24, 25, 28, 29, 30, 31, 32 | (3), (23), 24, 25, (26), 32 | 8, (9), 31 |
| B2 + B1 | 1, 2, 3, 4, 5, 6, 8, 10, 11, 12, 13, 14, 15, 16, 24, 25, 28, 29, 30, 31, 32, 54, 55, 56, 58 | 3, (23), 24, 25, (26), 32 | 8, (9), 10, 11, 12, 13, 14, 31, 55 |
| Class | Cluster | NativeP-PContacts |
| 429, 432, 433, 436, 439, 444, 445, 446, 447, 449, 450, 451, 459, 463 | ||
| D3 | 443, 444, 445, 446, 447, 448, 449, 451 | 444, 445, 446, 447, 449, (450), 451 |
| D3 + D2 | 436, 437, 442, 443, 444, 445, 446, 447, 448, 449, 450, 451, 452, 453, 456 | 436, 444, 445, 446, 447, 449, 450, 451 |
| B2 | 411, 412, 413, 414, 415, 416, 417, 418, 419, 420, 421, 422, 423, 424, 425, 426, 427, 428, 429, 430, 466, 470, 477 | 429 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banach, M.; Chomilier, J.; Roterman, I. Contribution to the Understanding of Protein–Protein Interface and Ligand Binding Site Based on Hydrophobicity Distribution—Application to Ferredoxin I and II Cases. Appl. Sci. 2021, 11, 8514. https://doi.org/10.3390/app11188514
Banach M, Chomilier J, Roterman I. Contribution to the Understanding of Protein–Protein Interface and Ligand Binding Site Based on Hydrophobicity Distribution—Application to Ferredoxin I and II Cases. Applied Sciences. 2021; 11(18):8514. https://doi.org/10.3390/app11188514
Chicago/Turabian StyleBanach, Mateusz, Jacques Chomilier, and Irena Roterman. 2021. "Contribution to the Understanding of Protein–Protein Interface and Ligand Binding Site Based on Hydrophobicity Distribution—Application to Ferredoxin I and II Cases" Applied Sciences 11, no. 18: 8514. https://doi.org/10.3390/app11188514