Abstract
Multiple sclerosis (MS) is common neurological disease of the central nervous system (CNS) affecting mostly young adults. Despite decades of studies, its etiology and pathogenesis are not fully unraveled and treatment is still insufficient. The vast majority of studies suggest that the immune system plays a major role in MS development. This is also supported by the effectiveness of currently available MS treatments that target immunocompetent cells. In this review, the role of antigen-presenting cells (APC) in MS development as well as the novel therapeutic options targeting those cells in MS are presented. It is known that in MS, peripheral self-antigen-specific immune cells are activated during antigen presentation process and they enter the CNS through the disrupted blood–brain barrier (BBB). Myelin-reactive CD4+ T-cells can be activated by dendritic cells, infiltrating macrophages, microglia cells, or B-cells, which all express MHC class II molecules. There are also suggestions that brain endothelial cells may act as non-professional APCs and present myelin-specific antigens with MHC class II. Similarly, astrocytes, the major glial cells in the CNS, were shown to act as non-professional APCs presenting myelin antigens to autoreactive T-cells. Several currently available MS drugs such as natalizumab, fingolimod, alemtuzumab, and ocrelizumab may modulate antigen presentation in MS. Another way to use this mechanism in MS treatment may be the usage of specific tolerogenic dendritic cells or the induction of tolerance to myelin antigens by peptide vaccines.
1. Introduction
Multiple sclerosis (MS) is an autoimmune, inflammatory, and demyelinating disease of the central nervous system (CNS). Its etiology is still unknown, despite the fact that the new concepts appear with more in-depth studies. The pathogenesis is clearer, and we know that pathological processes such as inflammation, demyelination, astrogliosis, and neurodegeneration play an important role in this complex mechanism [1]. For many years, autoimmune inflammation was considered to be the initial step in MS pathogenesis. It is suggested that the major target of autoimmune attack is protein autoantigens localized in the CNS myelin sheaths. This concept was confirmed in animal models of the disease [2]. The complex clinical and pathological picture of MS is not reflected by single animal models. Rather, various animal models are utilized for studies on different aspects of this disease. One of the most frequently used model, especially in the context of autoimmunity, is EAE (experimental autoimmune encephalomyelitis). There are two main types of EAE—active and passive—depending on the selected method of induction [3]. Moreover, this model can be induced in various animals, such as mice, rats, guinea pigs, and primates. EAE resembles MS in pathological changes observed in the CNS (inflammation, demyelination, neurodegeneration) and in some aspects of clinical signs [4]. However, the etiology has varied greatly, as EAE is induced by animal immunization with selected myelin peptides, whereas in patients with MS, the autoantigen responsible for disease induction is not known.
The clinical picture of MS is also complex. Four different clinical courses of MS can be distinguished. The most common (85%) is the remitting–relapsing type of the disease characterized by the appearance of neurological attacks of the disease. Those attacks are clinical manifestations of acute inflammatory events in the CNS and last at least 24 h [5]. The type of the symptoms depends on the localization of the lesion in the CNS. Generally, those symptoms can be visual, motor, sensory, or cognitive. Relapses are separated by the spontaneous remissions, which are the consequence of healing processes in the CNS. The duration of the remissions is unpredictable and may last weeks, months, or even years. Approximately half of the patients with remitting–relapsing MS will develop the secondary progressive form of MS after 10 years [6]. This is the critical point of the disease, because it represents the situation where neurodegeneration starts to dominate in the CNS over inflammation. In some patients, this situation is present from the beginning of the disease and this is the most serious third clinical course of MS called primary progressive MS. In this variant, the disease constantly progresses without any remissions. The last clinical type of MS is the combination of previous ones whereby the patients experience relapses and progression of the disease. This form is called progressive relapsing MS [7]. Progressive forms of MS are bad predictors for the patients future, because the vast majority of currently available drugs for MS are registered for remitting–relapsing MS. Those treatments target the patient immune system. So far, there are no effective drugs for the treatment of neurodegeneration predominating in progressive MS.
Treatment of MS can be divided into: (a) treatment of MS symptoms, (b) treatment of MS relapse, and (c) treatment modifying disease progression. The major goal of MS treatment is slowing down the disease progression. Several medicines have shown to be effective in this area during the last twenty years. The first drugs were beta interferons and glatiramer acetate injected s.c. or i.m.; later, i.v (natalizumab) and p.o. drugs (fingolimod) were developed. The newer medicines are more effective than the previous ones to diminish relapse rate and slow down MS progression, but they also ameliorate pathological changes seen in MRI of MS patients. The mechanisms of action of disease-modifying treatments in MS are varied. Some drugs target the migration of inflammatory cells (natalizumab, fingolimod), others target specific subpopulations of immunocompetent cells important for the pathogenesis of MS, but most of the medicines used in MS have very complex mechanisms of action [3].
There is no doubt that MS and EAE are complex autoimmune disorders. In its complexity, MS involves the interplay of many immune processes, immune cells, and also environmental and genetic factors, which contribute to the risk of disease onset and its clinical course. Each of these factors has already been widely discussed and undergoes constant investigations; however, there are still many unsolved or unanswered questions regarding the MS issue, as well as new aspects of cellular activity have become a matter of debates. Nowadays, antigen-presenting cells and their therapeutic potential are in the spotlight of the researchers’ and clinicians’ interest.
2. The Role of Antigen-Presenting Cells in MS Pathogenesis
The pathobiology of MS is complex, and despite intense studies, the initial events leading to autoimmunity development have still not been unraveled. However, it is known that peripheral self-antigen-specific immune cells are activated during the antigen presentation process and they enter the CNS through the disrupted blood–brain barrier (BBB), the subarachnoid space, or the blood–CSF barrier [8,9,10,11]. The route of entry depends on the phenotype and activation state of T-cells [12]. After transmigration into the CNS, myelin-specific T-cells are re-activated by CNS-resident APCs (dendritic cells, microglia), which results in the development and/or exacerbation of inflammatory reaction and demyelination [13,14].
Antigen-presenting cells convert myelin antigens to various epitopes and present them with MHC class I and/or II molecules to the T-cell receptors (TCRs) of CD4 and CD8 cells. The activation of protease synthesis and antigen processing as well as synthesis of functional MHC molecules are indispensable signal mechanisms [15]. Additionally, for successful antigen presentation during T-cell priming and re-activation, other co-stimulatory molecules such as CD40, CD80, CD86, and integrins are required [16,17].
Myelin-reactive CD4+ T-cells are activated by dendritic cells, microglia cells, infiltrating macrophages, and B-cells—all expressing MHC class II molecules. CD8+ T-cells, which are highly present in the acute lesions of MS, mediate inflammation and demyelination after their activation by myelin peptide presentation with APCs expressing MHC class I molecules. All nucleated cells, including oligodendrocytes, astrocytes, and neurons, expressed MHC I [18,19,20].
The antigen-priming of T-cells is an important process in chronic and acute inflammation. In animal models of MS, infiltrating auto-reactive CD4+ T-cells are re-activated within CNS by APCs, which results in monocyte recruitment into the CNS, as well as in naïve CD4+ T-cells priming through epitope spreading, which boosts disease inflammatory reaction.
In autoimmune diseases, epitope spreading contributes to chronicity of inflammation and to diversification of the ongoing immune response. Epitope spreading is a process in which immune responses are directed to epitopes that vary from the initial ones [9]. In EAE, it has been shown that initial immune response is focused on a certain epitope and, then, during disease progression, spreads to others [21,22]. Spreading of epitopes may be intramolecular—within one molecule, e.g., various epitopes of MBP, or intermolecular, e.g., from PLP to MOG [21,23,24]. Various EAE models suggested that this process may begin within CNS, and that it was associated with clinical relapses [24,25]. Both intramolecular and intermolecular epitope spreading has also been observed in patients with MS [26,27,28]. However, what is the exact role of such process on disease course requires further analyses, as some studies were not able to detect any associations with disease exacerbations [26,29].
Numerous studies emphasize the importance of microglia and infiltrated dendritic cells/macrophages as an antigen-presenting cells in the CNS. Recently, it was speculated that B-cells may also act as professional APCs.
3. Dendritic Cells
A lot has been said about the pathogenesis of MS and the role of T-cells in inflammation and demyelination of the CNS. Considering MS as an autoimmune disorder, it could be assumed that initiation of immune reaction is linked with an interplay between the elements of innate and adoptive immunity. As the disease is characterized by a specific immune response toward the CNS myelin antigens, its induction depends on the activation of naive T-cells recognizing processed myelin antigens. Various cell types are able to present antigens for T-lymphocytes; however, only dendritic cells are called the professional antigen-presenting cells—it means that only these cells are capable of priming naive T-cells to differentiation. Since their first description by Steinman [30,31,32,33], dendritic cells became considered one of the most important players in the induction of antigen-specific immune responses. Actually, dendritic cells are the aim of newly designed therapeutic strategies in every field where the specific immune response matters (i.e., allergic disorders, cancer therapies, autoimmune disorders) [34].
Because of their biological properties, DCs are thought to be a key players in the initiation of MS, its development, maintenance, and progression. Dendritic cells are heterogeneous group of cells localized in various tissues, where they act as sentinels—filtrating extracellular tissue environment for antigens. In this functional state, dendritic cells possess immature phenotype due to their low ability for antigen presentation, but high endocytic activity and high expression of molecules that facilitates antigen recognition and uptake, for example C-type lectin mannose receptor MR (CD206) or DC-SIGN (CD209). Ingested antigens are processed and presented on the MHC molecules class II (but also class I) on the cell surface. For efficient antigen presentation, antigen uptake must be associated with dendritic cell maturation—which is triggered by danger signals provided with foreign (bacterial or viral) antigens and recognized by DCs via the set of pattern recognition receptors recognizing pathogen-associated molecular patterns [35]. Danger signals may also be provided by tissue destruction and inflammatory cytokines released from other cells. Depending on the recognized signal, located in tissue, immature DCs undergo maturation and their phenotype changes: the expression of antigen uptake receptors as the pinocytic and endocytic activity decreases, whereas the expression of MHC class II and co-stimulatory molecules (CD86, CD83, CD80, CD40) increases. During the maturation, DCs migrate from the tissue via lymph vessels to the draining lymph nodes. Migration to the lymph nodes is regulated by the chemokine milieu. The expression of CCR7 molecules on the surface of maturing DCs regulates their trafficking to the lymph nodes in response to CCL19 and CC21 chemokines [36,37]. This chemotactic process is also regulated by CXCR4—a receptor for chemokine CXCL12 [38]. In lymph nodes, mature DCs present processed antigens for T-cells, prime their activation, and shape the development of antigen-specific immune response. If the migrated DCs are not activated enough, then the possible result of antigen presentation for T-cells will be the anergy of antigen-specific T-cells or induction of Treg cells, resulting in the induction of peripheral tolerance to antigens. Recent findings suggest that the ability of DCs to prime immune tolerance to presented antigen is linked rather with a specific activation state developed after antigen recognition (tolerogenic phenotype) than the lack of maturation. The maturation process occurs, however, into the different phenotype.
In MS, the uptake and transport of brain antigens by DCs or another APC cells has been contentious, due to the alleged independence of CNS from the immune system and the presence of BBB, which was believed to block immune cells trafficking into the brain. Recent findings have finally shown that dendritic cells may also be present in the central nervous system not only during inflammation, but also at steady-state conditions. It is discussible whether DCs may home the brain parenchyma; however, it is already confirmed that these cells are present in choroid plexus, perivascular spaces of BBB, meninges, and in cerebrospinal fluid. Their presence seems to be the effect of migration of bone marrow precursors from blood [39]. It was also found that microglia cells—brain-specific macrophages—may differentiate into immature dendritic cells after GM-CSF stimulation [40]; however, the comparative microarray analysis of gene expression profile suggests the close resemblance of the brain DCs to the dendritic cells of spleen, but not to the microglia [41]. Localization of DCs in close proximity to neurons gives a chance for myelin antigens uptake and initiation disease-specific immune response. The myelin antigens uptake and the source of danger signals are still open and unsolved questions. It seems rather unbelievable that myelin antigens alone may provide the efficient signal for DCs’ activation and maturation; however, coexisting viral infections as well as local physical injuries may strengthen the danger signal.
Another aspect of specific immune response is the place of naive T-cells priming by antigen-loaded mature DCs. On periphery, antigen-processing DCs migrate to the draining lymph nodes via lymphatic vessels. However, there is no typical lymphatic drainage of the brain and the exact path for dendritic cells’ migration is not yet fully identified. However, observations on animal models describe the presence of labeled OVA antigens in cervical lymph nodes after its intracerebral injection into the brain [42]. Accumulation of myelin-loaded CD11c-positive dendritic cells in cranial lymph nodes was also reported [43]. Studies on dendritic cells loaded with antigens and injected into the cerebellum or CSF also reported their movement to the cervical lymph nodes and localized their antigen presentation [44,45]. The exact route for cells migration from brain to draining lymph nodes is not fully identified. Recent studies describe the role of meningeal lymphatic vessels as a route for antigen-loaded cells from the brain to the cranial lymph nodes [46,47,48]. Another route for antigen-presenting cells from the brain to the peripheral lymph nodes was reported by Hochmeister et al., where injection of monocyte-derived DCs into the striatum resulted in their perivascular accumulation and migration across the endothelium into the vessel lumen [49]. The third possible route for CD11c-positive DCs was reported by Mohammad et al. and utilizes olfactory bulb and rostral migratory stream [50].
The separate issue is the possible role of microglia cells as potential professional APCs and their possible role in initiation of antigen-specific immune response in MS. Microglia are cells that are located in the brain parenchyma in the close proximity to myelin. The recent findings of Schiefenhövel et al. suggest the possible ability of microglia cells to migrate from the brain parenchyma to cervical lymph nodes for antigen presentation [51].
Microglia represents a subset of CNS resident cells with macrophage-like morphology, derived from early post-embryonic precursors. In resting state, ramified microglia continually surveys the microenvironment for threats. The activated amoeboid microglia have round or oval shape, process antigens, and clear them from the vicinity. Microglia may be divided into two subgroups: M1, which promote inflammation and oligodendrocyte damage, and M2, which regulate immune function, clear cellular debris, and promote repair in inflammatory diseases of the CNS [52,53]. However, now, it is assumed that microglia may differentiate into various subtypes with diverse functions, which is affected by a variety of environmental stimuli [54,55,56,57,58].
Microglia express MHC class I and II molecules and secrete many pro- and anti-inflammatory cytokines, as well as co-stimulatory molecules, such as intercellular adhesion molecule-1 (ICAM-1), CD80, and CD86 [59,60]. As APCs, microglia interact with other immune cells, which leads to the activation of various T-cell subsets during demyelination and remyelination [59,61]. Microglia can interact with CD28 or CTLA4 expressed on T-cells, which results in different outcomes. Microglial co-stimulatory molecules CD80 and CD86 bind CD28 to stimulate T-cells proliferation, differentiation, and cytokine production. Conversely, binding to CTLA4 leads to T-cell anergy or apoptosis [62].
Results from a number of studies indicated that microglia are not effective as APCs, DCs, or macrophages [8,63,64]. However, contradictory results exist. Wlodarczyk et al. have shown that DC and microglia sorted from CNS of mice with EAE have similar expression levels of MHC class I and II, CD80, and CD86. Moreover, it was shown that both cell populations were able to induce an antigen-specific proliferative response in primed T-cells. However, CD11c+ microglia were weak inducers of Th1 and Th17 differentiation, due to the lack of expression of necessary cytokines, conversely to infiltrating CD11c+ cells that strongly induced such cytokines [65]. Interestingly, CD11c− microglia have been shown to be poor inducers of T-cells proliferation, but this subset has the capability to produce Th1- and Th17-inducing cytokines, pointing to the synergized mode of action of both microglia subpopulations [65]. Thus, it is speculated that CD11c+ and CD11c− microglia may function as CNS-resident APCs being as effective as infiltrating CD11c+ cells.
B-cells, despite their function in antibodies production, may also act as antigen-presenting cells. It has been demonstrated that in the subset of MS patients, B-cells supported proliferation and/or IFN-γ production by autologous T-cells in response to neuroantigens, such as MOG and MBP [66]. Additionally, B-cells from MS patients, but not from healthy donors (HD), were able to induce proliferation and IL-17 production by Th17 cells in response to neuroantigens [67]. Similar results were obtained in the EAE model, where B-cells also promoted MOG-specific Th1 and Th17 cell differentiation [68]. Additionally, animals with MHC class II molecules deficiencies were resistant to disease induction and have attenuated Th1 and Th17 responses [69].
Several studies have shown that CD40-activated B-cells from HD and MS patients induce T-cell responses specific to myelin antigens [70,71]. However, Ireland et al. have indicated that B-cells from MS patients may support myelin antigen-specific T-cell priming without previous in vitro activation [67]. Thus, B-cells are efficient APCs, capturing antigens through a membrane-bound B-cell receptor, processing them, and presenting via MHC class II molecules [72]. Moreover, it has been reported that B-cells from the peripheral blood of MS patients have increased the expression of co-stimulatory CD40 and CD80 molecules compared to HD [73]. Immunomodulatory treatment decreased B-cell co-stimulatory molecules expression, resulting in reduced T-cell responses induced by B-cells [74,75]. The increased expression level of co-stimulatory molecules CD80, CD86, and HLA-A/B/C was also observed on B-cells isolated from CSF [73].
Clonal expansion of B-cells allows them to activate many T-cells, thus leading to an exacerbation of inflammation. In an animal model of MS depending on B- and T-cells, it was shown that B-cell function as APCs is necessary for disease induction, instead of their ability to produce antibodies [69,72].
The vascular endothelial cells separating the blood stream from the brain parenchyma are referred to as a blood–brain barrier. This barrier regulates the entry of various substances and myeloid cells into the CNS, thus providing anatomical and physiological protection. Migration of myelin-specific CD4+ T-cells across the BBB, a crucial step in the pathogenesis of MS, is suggested to be an antigen-specific process. It has been proposed that this process is mediated by brain endothelium. Lopes et al. reported that in inflammatory conditions, brain endothelial cells (BECs) act as a non-professional APCs able to process and present myelin-derived antigens complexed with MHC class II. Such complexes stimulated myelin-reactive Th1 and Th17 2D2 cells to transmigration through endothelium. Blocking of interactions between myelin/MHC II complexes and reactive T-cells abrogated the trafficking of the latter across the BBB [11]. It has been reported that BECs express MHC class I, but the MHC class II is present in very low levels in physiological conditions. Induction of the inflammatory process activates BECs and results in increased expression of MHC II, CD40, and ICOSL and, subsequently, enhances their ability to induce T-cells proliferation in vitro [76]. Additionally, it was shown that myelin enters the endosomal/lysosomal pathway after internalization by BECs, which was autonomous from their activation status [77]. This is opposite to what is observed for professional APCs such as DCs, where internalization of antigens takes place in an immature state of these cells [78,79]. Overall, these results pointed out that brain endothelium is an active and important contributor to the pathogenesis of MS.
Astrocytes are the most abundant cells in the CNS and they can act as a non-professional APCs modulating the activity of autoreactive T-cells by myelin antigen presentation and secretion of pro- or anti-inflammatory cytokines.
The role of astrocytes as APCs has remained controversial. There are reports that have shown that astrocytes express low levels of MHC class II molecules constitutively, and their expression may be upregulated during inflammatory reactions [80,81]. The expression of co-stimulatory molecules, such as CD40, CD80, and CD86, is also upregulated during astrocytes activation [81,82,83]. However, studies utilizing human fetal astrocytes in vitro failed to detect the expression of co-stimulatory molecules, even after stimulation [84]. Functional studies have shown that stimulated murine astrocytes moderately activate CD4+ and CD8+ T-cells, whereas cytokine-treated human astrocytes did not induce proliferation of encephalitogenic T-cells [80]. These may reflect differences between species or artifacts associated with astrocytic cell cultures, which may be contaminated by microglia. It has also been reported that activated astrocytes efficiently presented MBP, PLP, and MOG epitopes [85,86,87]. Yet, they were incapable of processing and presenting native myelin peptides. Thus, the astrocytes potential for antigen presentation needs more detailed studies to elucidate its role for the development and progression of MS.
4. MS-Approved Drugs Targeting Immunocompetent Cells
Currently, several drugs used in MS therapy target inflammatory cell subpopulations, which play a significant role in MS pathogenesis (Table 1) [88]. Natalizumab and fingolimod are anti-migratory drugs and influence inflammatory T-cell migration to the CNS of MS patients. Natalizumab is a humanized monoclonal antibody targeting adhesion molecule α4-integrin on effector T-cells in the blood, which leads to the blockade of inflammatory cell migration to the CNS. This mechanism is very efficient leading to lowering of the annual relapse rate by 68% and diminishing the number of demyelinating lesions in MRI of the CNS by 92% [89]. Natalizumab is delivered by intravenous infusions every month. A very serious side effect of this treatment is the possibility of developing progressive multifocal leukoencephalopathy (PML), which may be limited by controlling the level of anti-JC antibody in candidate MS patients [90,91]. Fingolimod is the second anti-migratory drug used in MS and the first effective oral drug for this disease. It targets sphingosine S1P receptors. The major effect of this action is inhibition of migration of T-cells out of the lymphatic nodes. In consequence, effector T-cells cannot invade the CNS and initiate the development of MS pathology. This treatment effectively reduces annual relapse rate and slows down clinical and radiological disease progression. There are also solid data confirming positive influence of this treatment on brain atrophy in MS patients [92].

Table 1.
Immunomodulatory drugs for multiple sclerosis.
Alemtuzumab is recombinant humanized monoclonal antibody targeting glycoprotein CD52 on T and B-cells. It is delivered once a year, because drug infusions lead to the depletion of both types of lymphocytes, with their slow repopulation from non-affected progenitor cells. The mechanism of action of this drug is very complex. Besides T- and B-cell lymphopenia, another effect is an increased number of Tregs, upregulation of several inflammatory cytokines, and the induction of neurotrophin-producing lymphocytes. Alemtuzumab significantly inhibits the progression of MS symptoms and diminishes the annual relapse rate and number of new lesions in the CNS of MS patients [93,94].
The clinical trials on anti-CD20 mAbs therapeutic efficacy in MS revealed their significant potential in reducing the clinical and MRI activity in patients. Four currently studied mAbs—rituximab, ocrelizumab, ofatumumab, and ublituximab—differ from each other by their structure, immunogenicity (chimeric, humanized, fully human, or glycoengineered), the type of cytotoxicity they induce (antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity), and also by the CD20 epitope they recognize [95]. Studies on their mode of action in RR-MS patients suggested that the main mechanism is not related to a reduction in the Abs level, but it is rather the modulation of APC function of B-cells. B-cells, as was mentioned in the previous section, are able to process and present antigens to T-cells. They recognize myelin antigens in MS via their surface receptors, and they efficiently capture antigens presented at low concentration [96]. Moreover, CD20 is not expressed on plasmablasts or plasma cells, the B-cell subtypes responsible for antibody secretion. Thus, after CD20 mAbs treatment, there was no reduction observed in antibody titers [74,97]. It is worth mentioning that ocrelizumab is also the first disease-modifying therapy approved for primary progressive MS, but is also registered for patients with remitting–relapsing MS [98]. In multi-center randomized clinical trials, it demonstrated higher efficacy in both forms of MS, when compared to patients in the placebo group and group treated with interferon beta [99]. It reduces number of disease relapses, slows the progression of disability, and reduces number of MRI lesions in the CNS of MS patients [100].
Another example of surface receptor with therapeutical potential is CD83, especially its soluble form (sCD83). Recombinant soluble CD83 (rsCD83) have demonstrated its potential via immunosuppressive properties observed in animal models of autoimmune diseases, including MS [101]. All of the studies indicate that this receptor is able to suppress immune responses, via inhibition of human monocyte differentiation into DCs, changing the DC cytoskeleton, preventing DC maturation, and reducing DC-mediated T-cell proliferation [102,103,104,105,106,107]. Results obtained from EAE studies indicate that administration of rsCD83 prevents EAE development and also reduces the level of T-cell cytokines, such as IFN-γ, IL-2, IL-4 and IL-10 [101].
Dimethyl fumarate (DMF) is the first-line oral drug approved for RR-MS patients, with moderate to high efficacy [108]. The main mechanism of action of DMF is an alteration of Th-cells differentiation and total number, induction of regulatory B-cells, and its anti-oxidative potential [109]. The naïve and regulatory T-cell subpopulations are elevated, whereas there is a reduction in memory T-cells, as well as in Th1 and Th17 cells [110,111]. However, it was also shown that DMF may alter activity of APCs, such as monocytes and dendritic cells [108]. Moreover, it has been shown that DMF has ameliorated a B-cell-accentuated EAE by diminishing the capacity of B-cells to act as APCs for T-cells [112]. In the studies conducted on blood obtained from MS patients, it was reported that the total number of B-cells was reduced after DMF treatment, with the emphasis on differentiated cells [113,114,115,116]. The level of mature and memory B-cells, together with plasmablasts, was reduced, whereas the frequency of immature, transitional B-cells increased [112].
One of the therapeutic goals in MS treatment is the induction of long-term, durable, antigen-specific T-cell tolerance. Antigen-specific immunotherapies are emerging to suppress targeted immune responses without altering the global immune system [117,118].
4.1. Dendritic Cells in MS Therapy
As a key player in the initiation and maintenance of antigen-specific immune response, dendritic cells are considered not only as a target for drug-based treatment. The use of specific tolerogenic dendritic cells is also considered a therapeutic option. The idea of using tolerance-inducing dendritic cells depends on the use of antigen-loaded DCs primed to tolerogenic phenotype, which are able to polarize the immune response against presented antigens towards the induction of immune tolerance, which results in the expansion of antigen-specific regulatory T-cells. Phenotype of tolerance-inducing DCs is traditionally characterized as semi-mature with weak expression of MHC class II molecules and co-stimulatory receptors (CD86, CD83, CD80, CD40), high level of surface inhibitory molecules (PDL1/2, ILT3, ILT4), and specific secretory activity with evident IL-10 or TGF-β production and weak release of proinflammatory cytokines [122]. Induction of tolerance to antigens depends mainly on the ability of DCs to promote differentiation of naive T-cells to Treg during antigen presentation in lymph nodes. Moreover, tolerance-inducing DCs were also reported to be able to inhibit the proliferation of T-cells, induce T-cells hyporesponsiveness, and T-cells anergy [123,124,125]. The level of conventional DCs in circulating blood is quiet low; thus, in numerous studies, DCs are derived from blood monocytes. Standard protocol utilizes moDCs differentiation from monocytes in the presence of IL-4 and GM-CSF to immature DCs [126,127]. Tolerogenic phenotypes may be obtained in numerous ways; the most popular utilizes vitamin D3 or IL-10; however, the use of corticosteroids, rapamycin, or specific NF-κB inhibitors were also described as possible strategies [128].
Up to date, the most interesting and well-studied are tolerogenic DCs induced by vitamin D3 or glycocortycosteroid dexamethasone. In vitro and animal studies with vitD3-tolDC confirmed their ability to induce hyporesponsiveness of T-cells toward presented antigens, as well as the stability of promoted tolerogenic phenotype, which was not abolished by cryopreservation [129,130]. Animal studies conducted using an EAE model revealed the potential of myelin antigen-loaded tolDC administration to suppress immune response and enhance the proliferation of regulatory B-cells and T-cells. Recently, Mansilla et al. identified CD115 (CSF1R) as a biomarker for tolerogenic properties of DCs, which is also involved in the vitamin D-mediated induction of tolerogenic phenotypes. What is more, authors reported CSF1R-CSF1 signaling to be important for metabolic reprogramming of vitamin D-modulated DCs, related with elevated glucose uptake and lactate production [131]. Release of lactic acid is a novel, recently identified mechanism utilized by tolerogenic DCs to suppress T lymphocytes proliferation [132]. These findings improve further tolDC studies and improve time-consuming identification of tolerogenic phenotype of DCs induced with vitamin D. It should be checked if regulatory properties of another already described tolerance-inducing DCs, obtained on different priming protocols (dexamethasone, IL-10, TGF-β, synthetic NF-κB inhibitors), are also lactate and CSF1R-CSF1 signaling dependent. It is worth to consider CSF1R as a target for improvement of regulatory activity of antigen loaded DCs derived from monocytes, as well as circulating conventional DCs in the body.
A major disadvantage of this form of therapy is the necessity to use tolerogenic DCs loaded with multiple myelin antigens as the immune response among the patients is heterogeneous, and the antigen spreading must always be considered. Although experimental tolDC therapy was effective in early stages of EAE, there was no beneficial effect of its use for chronic EAE [129,133,134,135,136]. Similar results should be expected in accordance with MS in humans. In the early stage of inflammation, when the autoimmune reaction targets major myelin antigens, the tolDC-based therapy should be the most effective [136].
4.2. Tolerogenic DCs in Clinical Trials
Actually, two independent phase I clinical trials utilizing tolDC are taking place, and one has already finished [136,137]. A phase Ib clinical trial with dexamethasone-induced tolerogenic DC loaded with myelin antigens or Aquaporin 4 that finished in 2019 confirmed safety of the therapy and provided the proof of concept for use of tolerance inducing DCs in future. Within 12 weeks after intervention, a significant increase in IL-10 release by PBMC cells exposed to myelin antigens and elevated Treg frequency have been observed in study participants, which suggests the induction of immune tolerance to used antigens. What is important is that there was no serious side effects and during the 24-week follow-up, researchers noticed only non-severe episodes, not related with the study [138]. Such a form of therapy depending on restoring immune tolerance without general immune suppression is needed. Actually, two phase I studies are recruiting MS-affected participants to evaluate safety and tolerability of the treatment, as well as to determine the most suitable administration option of vitamin D-induced tolerogenic DC loaded with myelin immunopeptides (Figure 1). It seems necessary to evaluate the most effective route for tolDC administration, as well as the development of a fast and reliable method for the induction of tolerance-inducing DCs and assessment of the tolerogenic potential of DCs [138]. Future trials for testing the efficiency of tolDC-based therapies on the clinical level, involving a higher number of participants, should be carried out, as well as the frequency of side effects being evaluated.
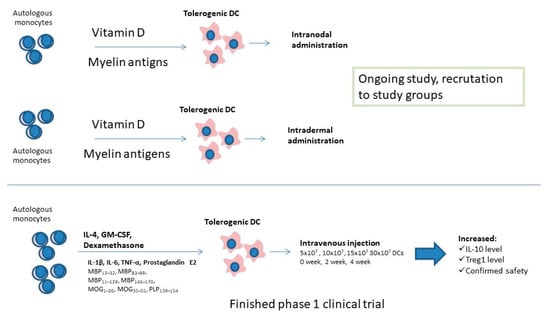
Figure 1.
Clinical studies conducted on tolerogenic dendritic cells.
4.3. Targeting the APC Activity by Peptide Vaccines
Induction of tolerance to myelin antigens may be also obtained by administration of the antigen by non-conventional route. Numerous trials investigated the effect of myelin antigens administered orally, intravenously, subcutaneously, or intranodally. Therapy with myelin peptides depends on their uptake without strong activation of APCs, which induce tolerance to the antigen. Trials utilizing the immune peptides administration were summarized in Table 2. Common disadvantages of peptide administration are the necessity of repetitive administration in high doses and the fast clearance of injected peptide [122,139,140,141].
Table 2.
Clinical trial results for myelin antigen use as a therapeutic options in multiple sclerosis. Trials without any beneficial effect are labeled with black shade; trials with strong side effects are labeled with red shade; trials with positive results are labeled in green.
This process may be facilitated by the incorporation of selected peptides into the liposomes modified to elevate their uptake by DCs via specific surface receptors. For example, immunodominant MBP peptides are incorporated into mannosylated liposomes, which increase their uptake by APCs via the CD206 receptor. This approach enhances immune tolerance for the CNS antigens [151,152].
Another option is nanoparticles loaded with self-antigen, with or without a bioactive payload, which have been developed to induce immune tolerance [153,154,155,156,157]. It has been shown that biodegradable particles loaded with myelin-specific antigen, either alone or with immunomodulators, ameliorate EAE [158,159]. Particles around 500 nm in size are phagocytosed by APCs [160,161,162]. Poly (lactide-co-glycolide) (PLG) and poly (DL-lactide) (PLA) particles have been mostly investigated in antigen-specific treatment for MS and have shown the capability to modulate immune cells for EAE disease amelioration [157,159,163].
5. Conclusions
Several currently available experimental data suggest that targeting antigen presentation may be a promising way to develop the new, more effective methods of MS treatment.
Author Contributions
P.S. and D.K.-W.—conceptualization, data search, writing—original draft; A.G.—conceptualization, writing—review and editing. P.S. and D.K.-W. contributed equally to this work. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by statutory research fund of Medical University of Lodz, Poland, 503/5-062-01/503-51-001-19-00.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare that they have no conflict of interest.
References
- Trapp, B.D.; Nave, K.-A. Multiple Sclerosis: An Immune or Neurodegenerative Disorder? Annu. Rev. Neurosci. 2008, 31, 247–269. [Google Scholar] [CrossRef]
- Korn, T.; Mitsdoerffer, M.; Kuchroo, V.K. Immunological Basis for the Development of Tissue Inflammation and Organ-Specific Autoimmunity in Animal Models of Multiple Sclerosis BT—Molecular Basis of Multiple Sclerosis: The Immune System; Martin, R., Lutterotti, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 43–74. ISBN 978-3-642-14153-9. [Google Scholar]
- Linker, R.A.; Lee, D.-H. Models of autoimmune demyelination in the central nervous system: On the way to translational medicine. Exp. Transl. Stroke Med. 2009, 1, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, C.; Strazielle, N.; Ghersi-Egea, J.-F. Brain leukocyte infiltration initiated by peripheral inflammation or experimental autoimmune encephalomyelitis occurs through pathways connected to the CSF-filled compartments of the forebrain and midbrain. J. Neuroinflamm. 2012, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Katz Sand, I. Classification, diagnosis, and differential diagnosis of multiple sclerosis. Curr. Opin. Neurol. 2015, 28, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Weinshenker, B.G.; Bass, B.; Rice, G.P.A.; Noseworthy, J.; Carriere, W.; Baskerville, J.; Ebers, G.C. The Natural History of Multiple Sclerosis: A Geographically Based Study: I. Clinical Course and Disability. Brain 1989, 112, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Ebers, G.C. Natural history of primary progressive multiple sclerosis. Mult. Scler. J. 2004, 10, S8–S15. [Google Scholar] [CrossRef]
- Goverman, J. Autoimmune T cell responses in the central nervous system. Nat. Rev. Immunol. 2009, 9, 393–407. [Google Scholar] [CrossRef] [Green Version]
- Riedhammer, C.; Weissert, R. Antigen Presentation, Autoantigens, and Immune Regulation in Multiple Sclerosis and Other Autoimmune Diseases. Front. Immunol. 2015, 6, 322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cepok, S.; Schreiber, H.; Hoffmann, S.; Zhou, D.; Neuhaus, O.; von Geldern, G.; Hochgesand, S.; Nessler, S.; Rothhammer, V.; Lang, M.; et al. Enhancement of Chemokine Expression by Interferon Beta Therapy in Patients With Multiple Sclerosis. Arch. Neurol. 2009, 66, 1216–1223. [Google Scholar] [CrossRef] [Green Version]
- Lopes Pinheiro, M.A.; Kooij, G.; Mizee, M.R.; Kamermans, A.; Enzmann, G.; Lyck, R.; Schwaninger, M.; Engelhardt, B.; de Vries, H.E. Immune cell trafficking across the barriers of the central nervous system in multiple scl/erosis and stroke. Biochim. Biophys. Acta-Mol. Basis Dis. 2016, 1862, 461–471. [Google Scholar] [CrossRef]
- Nishihara, H.; Soldati, S.; Mossu, A.; Rosito, M.; Rudolph, H.; Muller, W.A.; Latorre, D.; Sallusto, F.; Sospedra, M.; Martin, R.; et al. Human CD4+ T cell subsets differ in their abilities to cross endothelial and epithelial brain barriers in vitro. Fluids Barriers CNS 2020, 17, 1–18. [Google Scholar] [CrossRef]
- Mrdjen, D.; Pavlovic, A.; Hartmann, F.J.; Schreiner, B.; Utz, S.G.; Leung, B.P.; Lelios, I.; Heppner, F.L.; Kipnis, J.; Merkler, D.; et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 2018, 48, 380–395.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mundt, S.; Mrdjen, D.; Utz, S.G.; Greter, M.; Schreiner, B.; Becher, B. Conventional DCs sample and present myelin antigens in the healthy CNS and allow parenchymal T cell entry to initiate neuroinflammation. Sci. Immunol. 2019, 4, eaau8380. [Google Scholar] [CrossRef]
- Blum, J.S.; Wearsch, P.A.; Cresswell, P. Pathways of antigen processing. Annu. Rev. Immunol. 2013, 31, 443–473. [Google Scholar] [CrossRef] [Green Version]
- Szpakowski, P.; Biet, F.; Locht, C.; Paszkiewicz, M.; Rudnicka, W.; Druszczyńska, M.; Allain, F.; Fol, M.; Pestel, J.; Kowalewicz-Kulbat, M. Dendritic Cell Activity Driven by Recombinant Mycobacterium bovis BCG Producing Human IL-18, in Healthy BCG Vaccinated Adults. J. Immunol. Res. 2015, 2015, 359153. [Google Scholar] [CrossRef] [Green Version]
- Kowalewicz-Kulbat, M.; Szpakowski, P.; Locht, C.; Biet, F.; Kaplonek, P.; Krawczyk, K.T.; Pestel, J.; Rudnicka, W. Tuberculin skin test reaction is related to memory, but not naive CD4+ T cell responses to mycobacterial stimuli in BCG-vaccinated young adults. Vaccine 2018, 36, 4566–4577. [Google Scholar] [CrossRef] [PubMed]
- Babbe, H.; Roers, A.; Waisman, A.; Lassmann, H.; Goebels, N.; Hohlfeld, R.; Friese, M.; Schröder, R.; Deckert, M.; Schmidt, S.; et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J. Exp. Med. 2000, 192, 393–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobsen, M.; Cepok, S.; Quak, E.; Happel, M.; Gaber, R.; Ziegler, A.; Schock, S.; Oertel, W.H.; Sommer, N.; Hemmer, B. Oligoclonal expansion of memory CD8+ T cells in cerebrospinal fluid from multiple sclerosis patients. Brain 2002, 125, 538–550. [Google Scholar] [CrossRef]
- Hemmer, B.; Kerschensteiner, M.; Korn, T. Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol. 2015, 14, 406–419. [Google Scholar] [CrossRef]
- Klehmet, J.; Shive, C.; Guardia-Wolff, R.; Petersen, I.; Spack, E.G.; Boehm, B.O.; Weissert, R.; Forsthuber, T.G. T cell epitope spreading to myelin oligodendrocyte glycoprotein in HLA-DR4 transgenic mice during experimental autoimmune encephalomyelitis. Clin. Immunol. 2004, 111, 53–60. [Google Scholar] [CrossRef]
- Lehmann, P.V.; Forsthuber, T.; Miller, A.; Sercarz, E.E. Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature 1992, 358, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Mix, E.; Pahnke, J.; Ibrahim, S.M. Gene-Expression Profiling of Experimental Autoimmune Encephalomyelitis. Neurochem. Res. 2002, 27, 1157–1163. [Google Scholar] [CrossRef]
- Vanderlugt, C.L.; Neville, K.L.; Nikcevich, K.M.; Eagar, T.N.; Bluestone, J.A.; Miller, S.D. Pathologic Role and Temporal Appearance of Newly Emerging Autoepitopes in Relapsing Experimental Autoimmune Encephalomyelitis. J. Immunol. 2000, 164, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Carro, E.; Trejo, J.L.; Gomez-Isla, T.; LeRoith, D.; Torres-Aleman, I. Serum insulin-like growth factor I regulates brain amyloid-β levels. Nat. Med. 2002, 8, 1390–1397. [Google Scholar] [CrossRef]
- Ristori, G.; Giubilei, F.; Giunti, D.; Perna, A.; Gasperini, C.; Buttinelli, C.; Salvetti, M.; Uccelli, A. Myelin basic protein intramolecular spreading without disease progression in a patient with multiple sclerosis. J. Neuroimmunol. 2000, 110, 240–243. [Google Scholar] [CrossRef]
- Tuohy, V.K.; Yu, M.; Weinstock-Guttman, B.; Kinkel, R.P. Diversity and plasticity of self recognition during the development of multiple sclerosis. J. Clin. Investig. 1997, 99, 1682–1690. [Google Scholar] [CrossRef] [Green Version]
- Muraro, P.A.; Wandinger, K.; Bielekova, B.; Gran, B.; Marques, A.; Utz, U.; McFarland, H.F.; Jacobson, S.; Martin, R. Molecular tracking of antigen-specific T cell clones in neurological immune-mediated disorders. Brain 2003, 126, 20–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uccelli, A.; Ristori, G.; Giunti, D.; Seri, M.; Montesperelli, C.; Caroli, F.; Solaro, C.; Murialdo, A.; Marchese, M.; Buttinelli, C.; et al. Dynamics of the reactivity to MBP in multiple sclerosis. J. Neurovirol. 2000, 6 (Suppl. S2), S52–S56. [Google Scholar]
- Steinman, R.M.; Cohn, Z.A. Identification of A Novel Cell Type in Peripheral Lymphoid Organs of Mice: I. Morphology, Quantitation, Tissue Distribution. J. Exp. Med. 1973, 137, 1142–1162. [Google Scholar] [CrossRef]
- Steinman, R.M.; Cohn, Z.A. Identification of A Novel Cell Type in Peripheral Lymphoid Organs of Mice: II. Functional Properties in vitro. J. Exp. Med. 1974, 139, 380–397. [Google Scholar] [CrossRef] [Green Version]
- Steinman, R.M.; Lustig, D.S.; Cohn, Z.A. Identification of A Novel Cell Type in Peripheral Lymphoid Organs of Mice: III. Functional Properties in vivo. J. Exp. Med. 1974, 139, 1431–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinman, R.M.; Adams, J.C.; Cohn, Z.A. Identification of a novel cell type in peripheral lymphoid organs of mice. IV. Identification and distribution in mouse spleen. J. Exp. Med. 1975, 141, 804–820. [Google Scholar] [CrossRef] [PubMed]
- Sadeghzadeh, M.; Bornehdeli, S.; Mohahammadrezakhani, H.; Abolghasemi, M.; Poursaei, E.; Asadi, M.; Zafari, V.; Aghebati-Maleki, L.; Shanehbandi, D. Dendritic cell therapy in cancer treatment; the state-of-the-art. Life Sci. 2020, 254, 117580. [Google Scholar] [CrossRef] [PubMed]
- Patente, T.A.; Pinho, M.P.; Oliveira, A.A.; Evangelista, G.C.M.; Bergami-Santos, P.C.; Barbuto, J.A.M. Human Dendritic Cells: Their Heterogeneity and Clinical Application Potential in Cancer Immunotherapy. Front. Immunol. 2019, 9, 3176. [Google Scholar] [CrossRef]
- Hjortø, G.M.; Larsen, O.; Steen, A.; Daugvilaite, V.; Berg, C.; Fares, S.; Hansen, M.; Ali, S.; Rosenkilde, M.M. Differential CCR7 Targeting in Dendritic Cells by Three Naturally Occurring CC-Chemokines. Front. Immunol. 2016, 7, 568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britschgi, M.R.; Favre, S.; Luther, S.A. CCL21 is sufficient to mediate DC migration, maturation and function in the absence of CCL19. Eur. J. Immunol. 2010, 40, 1266–1271. [Google Scholar] [CrossRef]
- Ricart, B.G.; John, B.; Lee, D.; Hunter, C.A.; Hammer, D.A. Dendritic Cells Distinguish Individual Chemokine Signals through CCR7 and CXCR4. J. Immunol. 2011, 186, 53–61. [Google Scholar] [CrossRef] [Green Version]
- De Laere, M.; Berneman, Z.N.; Cools, N. To the Brain and Back: Migratory Paths of Dendritic Cells in Multiple Sclerosis. J. Neuropathol. Exp. Neurol. 2018, 77, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Fischer, H.-G.; Reichmann, G. Brain Dendritic Cells and Macrophages/Microglia in Central Nervous System Inflammation. J. Immunol. 2001, 166, 2717–2726. [Google Scholar] [CrossRef] [Green Version]
- Anandasabapathy, N.; Victora, G.D.; Meredith, M.; Feder, R.; Dong, B.; Kluger, C.; Yao, K.; Dustin, M.L.; Nussenzweig, M.C.; Steinman, R.M.; et al. Flt3L controls the development of radiosensitive dendritic cells in the meninges and choroid plexus of the steady-state mouse brain. J. Exp. Med. 2011, 208, 1695–1705. [Google Scholar] [CrossRef] [Green Version]
- Ling, C.; Sandor, M.; Fabry, Z. In situ processing and distribution of intracerebrally injected OVA in the CNS. J. Neuroimmunol. 2003, 141, 90–98. [Google Scholar] [CrossRef]
- Laman, J.D.; Weller, R.O. Drainage of cells and soluble antigen from the CNS to regional lymph nodes. J. Neuroimmune Pharmacol. 2013, 8, 840–856. [Google Scholar] [CrossRef]
- Karman, J.; Ling, C.; Sandor, M.; Fabry, Z. Initiation of Immune Responses in Brain Is Promoted by Local Dendritic Cells. J. Immunol. 2004, 173, 2353–2361. [Google Scholar] [CrossRef] [Green Version]
- Hatterer, E.; Davoust, N.; Didier-Bazes, M.; Vuaillat, C.; Malcus, C.; Belin, M.-F.; Nataf, S. How to drain without lymphatics? Dendritic cells migrate from the cerebrospinal fluid to the B-cell follicles of cervical lymph nodes. Blood 2006, 107, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.-X.; Liu, N.-K.; Wen, R.; Yang, S.-N.; Wen, X.; Xu, X.-M. Laminin-coated multifilament entubulation, combined with Schwann cells and glial cell line-derived neurotrophic factor, promotes unidirectional axonal regeneration in a rat model of thoracic spinal cord hemisection. Neural Regen. Res. 2021, 16, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Aspelund, A.; Antila, S.; Proulx, S.T.; Karlsen, T.V.; Karaman, S.; Detmar, M.; Wiig, H.; Alitalo, K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015, 212, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Herz, J.; Alme, M.N.; Salvador, A.F.; Dong, M.Q.; Viar, K.E.; Herod, S.G.; Knopp, J.; Setliff, J.C.; Lupi, A.L.; et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat. Neurosci. 2018, 21, 1380–1391. [Google Scholar] [CrossRef]
- Hochmeister, S.; Zeitelhofer, M.; Bauer, J.; Nicolussi, E.-M.; Fischer, M.-T.; Heinke, B.; Selzer, E.; Lassmann, H.; Bradl, M. After injection into the striatum, in vitro-differentiated microglia- and bone marrow-derived dendritic cells can leave the central nervous system via the blood stream. Am. J. Pathol. 2008, 173, 1669–1681. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, M.G.; Tsai, V.W.W.; Ruitenberg, M.J.; Hassanpour, M.; Li, H.; Hart, P.H.; Breit, S.N.; Sawchenko, P.E.; Brown, D.A. Immune cell trafficking from the brain maintains CNS immune tolerance. J. Clin. Invest. 2014, 124, 1228–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiefenhövel, F.; Immig, K.; Prodinger, C.; Bechmann, I. Indications for cellular migration from the central nervous system to its draining lymph nodes in CD11c-GFP+ bone-marrow chimeras following EAE. Exp. Brain Res. 2017, 235, 2151–2166. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.G.; Lue, L.-F. Immune phenotypes of microglia in human neurodegenerative disease: Challenges to detecting microglial polarization in human brains. Alzheimers. Res. Ther. 2015, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Kotter, M.R.; Setzu, A.; Sim, F.J.; Van Rooijen, N.; Franklin, R.J.M. Macrophage depletion impairs oligodendrocyte remyelination following lysolecithin-induced demyelination. Glia 2001, 35, 204–212. [Google Scholar] [CrossRef]
- Boche, D.; Perry, V.H.; Nicoll, J.A.R. Review: Activation patterns of microglia and their identification in the human brain. Neuropathol. Appl. Neurobiol. 2013, 39, 3–18. [Google Scholar] [CrossRef]
- Durafourt, B.A.; Moore, C.S.; Zammit, D.A.; Johnson, T.A.; Zaguia, F.; Guiot, M.-C.; Bar-Or, A.; Antel, J.P. Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia 2012, 60, 717–727. [Google Scholar] [CrossRef]
- Giunti, D.; Parodi, B.; Cordano, C.; Uccelli, A.; Kerlero de Rosbo, N. Can we switch microglia’s phenotype to foster neuroprotection? Focus on multiple sclerosis. Immunology 2014, 141, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Melief, J.; Koning, N.; Schuurman, K.G.; Van De Garde, M.D.B.; Smolders, J.; Hoek, R.M.; Van Eijk, M.; Hamann, J.; Huitinga, I. Phenotyping primary human microglia: Tight regulation of LPS responsiveness. Glia 2012, 60, 1506–1517. [Google Scholar] [CrossRef]
- Melief, J.; Schuurman, K.G.; van de Garde, M.D.B.; Smolders, J.; van Eijk, M.; Hamann, J.; Huitinga, I. Microglia in normal appearing white matter of multiple sclerosis are alerted but immunosuppressed. Glia 2013, 61, 1848–1861. [Google Scholar] [CrossRef]
- Frohman, E.M.; Racke, M.K.; Raine, C.S. Multiple Sclerosis—The Plaque and Its Pathogenesis. N. Engl. J. Med. 2006, 354, 942–955. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Jian, C.; Liao, Y.; Huang, Q.; Wu, Y.; Liu, X.; Zou, D.; Wu, Y. The role of microglia in multiple sclerosis. Neuropsychiatr. Dis. Treat. 2017, 13, 1661–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voß, E.V.; Škuljec, J.; Gudi, V.; Skripuletz, T.; Pul, R.; Trebst, C.; Stangel, M. Characterisation of microglia during de- and remyelination: Can they create a repair promoting environment? Neurobiol. Dis. 2012, 45, 519–528. [Google Scholar] [CrossRef]
- Bull, M.E.; Vahlenkamp, T.W.; Dow, J.L.; Collisson, E.W.; Winslow, B.J.; Phadke, A.P.; Tompkins, M.B.; Tompkins, W.A.F. Spontaneous T cell apoptosis in feline immunodeficiency virus (FIV)-infected cats is inhibited by IL2 and anti-B7.1 antibodies. Vet. Immunol. Immunopathol. 2004, 99, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Ford, A.L.; Foulcher, E.; Lemckert, F.A.; Sedgwick, J.D. Microglia induce CD4 T lymphocyte final effector function and death. J. Exp. Med. 1996, 184, 1737–1745. [Google Scholar] [CrossRef]
- Greter, M.; Heppner, F.L.; Lemos, M.P.; Odermatt, B.M.; Goebels, N.; Laufer, T.; Noelle, R.J.; Becher, B. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat. Med. 2005, 11, 328–334. [Google Scholar] [CrossRef]
- Wlodarczyk, A.; Løbner, M.; Cédile, O.; Owens, T. Comparison of microglia and infiltrating CD11c+ cells as antigen presenting cells for T cell proliferation and cytokine response. J. Neuroinflamm. 2014, 11, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Harp, C.T.; Ireland, S.; Davis, L.S.; Remington, G.; Cassidy, B.; Cravens, P.D.; Stuve, O.; Lovett-Racke, A.E.; Eagar, T.N.; Greenberg, B.M.; et al. Memory B cells from a subset of treatment-naïve relapsing-remitting multiple sclerosis patients elicit CD4+ T-cell proliferation and IFN-γ production in response to myelin basic protein and myelin oligodendrocyte glycoprotein. Eur. J. Immunol. 2010, 40, 2942–2956. [Google Scholar] [CrossRef]
- Ireland, S.J.; Guzman, A.A.; Frohman, E.M.; Monson, N.L. B cells from relapsing remitting multiple sclerosis patients support neuro-antigen-specific Th17 responses. J. Neuroimmunol. 2016, 291, 46–53. [Google Scholar] [CrossRef]
- Weber, M.S.; Prod’homme, T.; Patarroyo, J.C.; Molnarfi, N.; Karnezis, T.; Lehmann-Horn, K.; Danilenko, D.M.; Eastham-Anderson, J.; Slavin, A.J.; Linington, C.; et al. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann. Neurol. 2010, 68, 369–383. [Google Scholar] [CrossRef]
- Molnarfi, N.; Schulze-Topphoff, U.; Weber, M.S.; Patarroyo, J.C.; Prod’homme, T.; Varrin-Doyer, M.; Shetty, A.; Linington, C.; Slavin, A.J.; Hidalgo, J.; et al. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J. Exp. Med. 2013, 210, 2921–2937. [Google Scholar] [CrossRef]
- Arbour, N.; Lapointe, R.; Saikali, P.; McCrea, E.; Regen, T.; Antel, J.P. A new clinically relevant approach to expand myelin specific T cells. J. Immunol. Methods 2006, 310, 53–61. [Google Scholar] [CrossRef]
- Harp, C.T.; Lovett-Racke, A.E.; Racke, M.K.; Frohman, E.M.; Monson, N.L. Impact of myelin-specific antigen presenting B cells on T cell activation in multiple sclerosis. Clin. Immunol. 2008, 128, 382–391. [Google Scholar] [CrossRef]
- Lanzavecchia, A. Antigen-specific interaction between T and B cells. Nature 1985, 314, 537–539. [Google Scholar] [CrossRef] [PubMed]
- Fraussen, J.; Claes, N.; Van Wijmeersch, B.; van Horssen, J.; Stinissen, P.; Hupperts, R.; Somers, V. B cells of multiple sclerosis patients induce autoreactive proinflammatory T cell responses. Clin. Immunol. 2016, 173, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Cross, A.H.; Stark, J.L.; Lauber, J.; Ramsbottom, M.J.; Lyons, J.-A. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J. Neuroimmunol. 2006, 180, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Bar-Or, A.; Fawaz, L.; Fan, B.; Darlington, P.J.; Rieger, A.; Ghorayeb, C.; Calabresi, P.A.; Waubant, E.; Hauser, S.L.; Zhang, J.; et al. Abnormal B-cell cytokine responses a trigger of T-cell–mediated disease in MS? Ann. Neurol. 2010, 67, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Khaw, L.T.; Ball, H.J.; Golenser, J.; Combes, V.; Grau, G.E.; Wheway, J.; Mitchell, A.J.; Hunt, N.H. Endothelial Cells Potentiate Interferon-γ Production in a Novel Tripartite Culture Model of Human Cerebral Malaria. PLoS ONE 2013, 8, e69521. [Google Scholar] [CrossRef] [PubMed]
- Lopes Pinheiro, M.A.; Kamermans, A.; Garcia-Vallejo, J.J.; van het Hof, B.; Wierts, L.; O’Toole, T.; Boeve, D.; Verstege, M.; van der Pol, S.M.A.; van Kooyk, Y.; et al. Internalization and presentation of myelin antigens by the brain endothelium guides antigen-specific T cell migration. Elife 2016, 5, e13149. [Google Scholar] [CrossRef]
- Inaba, K.; Turley, S.; Iyoda, T.; Yamaide, F.; Shimoyama, S.; e Sousa, C.R.; Germain, R.N.; Mellman, I.; Steinman, R.M. The Formation of Immunogenic Major Histocompatibility Complex Class II–Peptide Ligands in Lysosomal Compartments of Dendritic Cells is Regulated by Inflammatory Stimuli. J. Exp. Med. 2000, 191, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Fuller, L.; Ciancio, G.; Burke, G.W.; Tzakis, A.G.; Ricordi, C.; Miller, J.; Esquenzai, V. Antigen presentation and immune regulatory capacity of immature and mature-enriched antigen presenting (dendritic) cells derived from human bone marrow. Hum. Immunol. 2004, 65, 93–103. [Google Scholar] [CrossRef]
- Weber, F.; Meinl, E.; Aloisi, F.; Nevinny-Stickel, C.; Albert, E.; Wekerle, H.; Hohlfeld, R. Human astrocytes are only partially competent antigen presenting cells: Possible implications for lesion development in multiple sclerosis. Brain 1994, 117, 59–69. [Google Scholar] [CrossRef]
- Cornet, A.; Bettelli, E.; Oukka, M.; Cambouris, C.; Avellana-Adalid, V.; Kosmatopoulos, K.; Liblau, R.S. Role of astrocytes in antigen presentation and naive T-cell activation. J. Neuroimmunol. 2000, 106, 69–77. [Google Scholar] [CrossRef]
- Zeinstra, E.; Wilczak, N.; De Keyser, J. Reactive astrocytes in chronic active lesions of multiple sclerosis express co-stimulatory molecules B7-1 and B7-2. J. Neuroimmunol. 2003, 135, 166–171. [Google Scholar] [CrossRef]
- Traugott, U. Multiple sclerosis: Relevance of Class I and Class II MHC-expressing cells to lesion development. J. Neuroimmunol. 1987, 16, 283–302. [Google Scholar] [CrossRef]
- Satoh, J.; Lee, Y.B.; Kim, S.U. T-cell costimulatory molecules B7-1 (CD80) and B7-2 (CD86) are expressed in human microglia but not in astrocytes in culture. Brain Res. 1995, 704, 92–96. [Google Scholar] [CrossRef]
- Soos, J.M.; Morrow, J.; Ashley, T.A.; Szente, B.E.; Bikoff, E.K.; Zamvil, S.S. Astrocytes express elements of the class II endocytic pathway and process central nervous system autoantigen for presentation to encephalitogenic T cells. J. Neuroimmunol. 1998, 161, 5959–5966. [Google Scholar] [CrossRef]
- Tan, L.; Gordon, K.B.; Mueller, J.P.; Matis, L.A.; Miller, S.D. Presentation of Proteolipid Protein Epitopes and B7-1-Dependent Activation of Encephalitogenic T Cells by IFN-γ-Activated SJL/J Astrocytes. J. Immunol. 1998, 160, 4271–4279. [Google Scholar]
- Kort, J.J.; Kawamura, K.; Fugger, L.; Weissert, R.; Forsthuber, T.G. Efficient presentation of myelin oligodendrocyte glycoprotein peptides but not protein by astrocytes from HLA-DR2 and HLA-DR4 transgenic mice. J. Neuroimmunol. 2006, 173, 23–34. [Google Scholar] [CrossRef]
- Rommer, P.S.; Zettl, R.P.; K Zettl, U. Monoclonal Antibodies in the Treatment of Neuroimmunological Diseases. Curr. Pharm. Des. 2012, 18, 4498–4507. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.H.; Khan, O.A.; Sheremata, W.A.; Blumhardt, L.D.; Rice, G.P.A.; Libonati, M.A.; Willmer-Hulme, A.J.; Dalton, C.M.; Miszkiel, K.A.; O’Connor, P.W. A Controlled Trial of Natalizumab for Relapsing Multiple Sclerosis. N. Engl. J. Med. 2003, 348, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Bloomgren, G.; Richman, S.; Hotermans, C.; Subramanyam, M.; Goelz, S.; Natarajan, A.; Lee, S.; Plavina, T.; Scanlon, J.V.; Sandrock, A.; et al. Risk of Natalizumab-Associated Progressive Multifocal Leukoencephalopathy. N. Engl. J. Med. 2012, 366, 1870–1880. [Google Scholar] [CrossRef]
- Coyle, P.K. The role of natalizumab in the treatment of multiple sclerosis. Am. J. Manag. Care 2010, 16, 164–170. [Google Scholar]
- Bermel, R.A.; Bakshi, R. The measurement and clinical relevance of brain atrophy in multiple sclerosis. Lancet Neurol. 2006, 5, 158–170. [Google Scholar] [CrossRef]
- Coles, A.J.; Twyman, C.L.; Arnold, D.L.; Cohen, J.A.; Confavreux, C.; Fox, E.J.; Hartung, H.-P.; Havrdova, E.; Selmaj, K.W.; Weiner, H.L.; et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: A randomised controlled phase 3 trial. Lancet 2012, 380, 1829–1839. [Google Scholar] [CrossRef]
- Cohen, J.A.; Coles, A.J.; Arnold, D.L.; Confavreux, C.; Fox, E.J.; Hartung, H.-P.; Havrdova, E.; Selmaj, K.W.; Weiner, H.L.; Fisher, E.; et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: A randomised controlled phase 3 trial. Lancet 2012, 380, 1819–1828. [Google Scholar] [CrossRef]
- Milo, R. Therapies for multiple sclerosis targeting B cells. Croat. Med. J. 2019, 60, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Linsley, P.S.; Clark, E.A.; Ledbetter, J.A. T-cell antigen CD28 mediates adhesion with B cells by interacting with activation antigen B7/BB-1. Proc. Natl. Acad. Sci. USA 1990, 87, 5031–5035. [Google Scholar] [CrossRef] [Green Version]
- Naismith, R.T.; Piccio, L.; Lyons, J.A.; Lauber, J.; Tutlam, N.T.; Parks, B.J.; Trinkaus, K.; Song, S.K.; Cross, A.H. Rituximab add-on therapy for breakthrough relapsing multiple sclerosis: A 52-week phase II trial. Neurology 2010, 74, 1860–1867. [Google Scholar] [CrossRef] [Green Version]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.-P.; Hemmer, B.; et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N. Engl. J. Med. 2016, 376, 209–220. [Google Scholar] [CrossRef]
- Kappos, L.; Li, D.; Calabresi, P.A.; O’Connor, P.; Bar-Or, A.; Barkhof, F.; Yin, M.; Leppert, D.; Glanzman, R.; Tinbergen, J.; et al. Ocrelizumab in relapsing-remitting multiple sclerosis: A phase 2, randomised, placebo-controlled, multicentre trial. Lancet 2011, 378, 1779–1787. [Google Scholar] [CrossRef]
- Hauser, S.L.; Bar-Or, A.; Comi, G.; Giovannoni, G.; Hartung, H.-P.; Hemmer, B.; Lublin, F.; Montalban, X.; Rammohan, K.W.; Selmaj, K.; et al. Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2016, 376, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Zinser, E.; Lechmann, M.; Golka, A.; Lutz, M.B.; Steinkasserer, A. Prevention and Treatment of Experimental Autoimmune Encephalomyelitis by Soluble CD83. J. Exp. Med. 2004, 200, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Zhu, Y.; Zhang, G.; Gao, C.; Zhong, W.; Zhang, X. CD83-stimulated monocytes suppress T-cell immune responses through production of prostaglandin E2. Proc. Natl. Acad. Sci. USA 2011, 108, 18778–18783. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Liang, S.; Zhong, Z.; Wen, J.; Li, W.; Wang, L.; Xu, J.; Zhong, F.; Li, X. Soluble CD83 inhibits human monocyte differentiation into dendritic cells in vitro. Cell. Immunol. 2014, 292, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Kotzor, N.; Lechmann, M.; Zinser, E.; Steinkasserer, A. The soluble form of CD83 dramatically changes the cytoskeleton of dendritic cells. Immunobiology 2004, 209, 129–140. [Google Scholar] [CrossRef]
- Lechmann, M.; Krooshoop, D.J.E.B.; Dudziak, D.; Kremmer, E.; Kuhnt, C.; Figdor, C.G.; Schuler, G.; Steinkasserer, A. The Extracellular Domain of CD83 Inhibits Dendritic Cell–mediated T Cell Stimulation and Binds to a Ligand on Dendritic Cells. J. Exp. Med. 2001, 194, 1813–1821. [Google Scholar] [CrossRef]
- Bates, J.M.; Flanagan, K.; Mo, L.; Ota, N.; Ding, J.; Ho, S.; Liu, S.; Roose-Girma, M.; Warming, S.; Diehl, L. Dendritic cell CD83 homotypic interactions regulate inflammation and promote mucosal homeostasis. Mucosal Immunol. 2015, 8, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Senechal, B.; Boruchov, A.M.; Reagan, J.L.; Hart, D.N.J.; Young, J.W. Infection of mature monocyte-derived dendritic cells with human cytomegalovirus inhibits stimulation of T-cell proliferation via the release of soluble CD83. Blood 2004, 103, 4207–4215. [Google Scholar] [CrossRef] [Green Version]
- Gold, R.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M.T.; Yang, M.; Sheikh, S.I.; et al. Placebo-Controlled Phase 3 Study of Oral BG-12 for Relapsing Multiple Sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmen, A.; Gold, R. Mode of action and clinical studies with fumarates in multiple sclerosis. Exp. Neurol. 2014, 262, 52–56. [Google Scholar] [CrossRef]
- Wu, Q.; Wang, Q.; Mao, G.; Dowling, C.A.; Lundy, S.K.; Mao-Draayer, Y. Dimethyl Fumarate Selectively Reduces Memory T Cells and Shifts the Balance between Th1/Th17 and Th2 in Multiple Sclerosis Patients. J. Immunol. 2017, 1601532. [Google Scholar] [CrossRef] [Green Version]
- Longbrake, E.E.; Ramsbottom, M.J.; Cantoni, C.; Ghezzi, L.; Cross, A.H.; Piccio, L. Dimethyl fumarate selectively reduces memory T cells in multiple sclerosis patients. Mult. Scler. J. 2015, 22, 1061–1070. [Google Scholar] [CrossRef] [Green Version]
- Traub, J.; Traffehn, S.; Ochs, J.; Häusser-Kinzel, S.; Stephan, S.; Scannevin, R.; Brück, W.; Metz, I.; Weber, M.S. Dimethyl fumarate impairs differentiated B cells and fosters central nervous system integrity in treatment of multiple sclerosis. Brain Pathol. 2019, 29, 640–657. [Google Scholar] [CrossRef] [Green Version]
- Høglund, R.A.; Polak, J.; Vartdal, F.; Holmøy, T.; Lossius, A. B-cell composition in the blood and cerebrospinal fluid of multiple sclerosis patients treated with dimethyl fumarate. Mult. Scler. Relat. Disord. 2018, 26, 90–95. [Google Scholar] [CrossRef]
- Lundy, S.K.; Wu, Q.; Wang, Q.; Dowling, C.A.; Taitano, S.H.; Mao, G.; Mao-Draayer, Y. Dimethyl fumarate treatment of relapsing-remitting multiple sclerosis influences B-cell subsets. Neurol.-Neuroimmunol. Neuroinflamm. 2016, 3, e211. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.D.; Martin, K.A.; Calabresi, P.A.; Bhargava, P. Dimethyl fumarate alters B-cell memory and cytokine production in MS patients. Ann. Clin. Transl. Neurol. 2017, 4, 351–355. [Google Scholar] [CrossRef]
- Spencer, C.M.; Crabtree-Hartman, E.C.; Lehmann-Horn, K.; Cree, B.A.C.; Zamvil, S.S. Reduction of CD8+ T lymphocytes in multiple sclerosis patients treated with dimethyl fumarate. Neurol.-Neuroimmunol. Neuroinflamm. 2015, 2, e76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishimoto, T.K.; Maldonado, R.A. Nanoparticles for the Induction of Antigen-Specific Immunological Tolerance. Front. Immunol. 2018, 9, 230. [Google Scholar] [CrossRef] [PubMed]
- Northrup, L.; Christopher, M.A.; Sullivan, B.P.; Berkland, C. Combining antigen and immunomodulators: Emerging trends in antigen-specific immunotherapy for autoimmunity. Adv. Drug Deliv. Rev. 2016, 98, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Lammens, A.; Schäfer, W.; Georges, G.; Schwaiger, M.; Mössner, E.; Hopfner, K.-P.; Umaña, P.; Niederfellner, G. Epitope interactions of monoclonal antibodies targeting CD20 and their relationship to functional properties. MAbs 2013, 5, 22–33. [Google Scholar] [CrossRef] [Green Version]
- De Jong, R.; Bezemer, A.C.; Zomerdijk, T.P.L.; van de Pouw-Kraan, T.; Ottenhoff, T.H.M.; Nibbering, P.H. Selective stimulation of T helper 2 cytokine responses by the anti-psoriasis agent monomethylfumarate. Eur. J. Immunol. 1996, 26, 2067–2074. [Google Scholar] [CrossRef]
- Yadav, S.K.; Soin, D.; Ito, K.; Dhib-Jalbut, S. Insight into the mechanism of action of dimethyl fumarate in multiple sclerosis. J. Mol. Med. 2019, 97, 463–472. [Google Scholar] [CrossRef]
- Moorman, C.D.; Sohn, S.J.; Phee, H. Emerging Therapeutics for Immune Tolerance: Tolerogenic Vaccines, T cell Therapy, and IL-2 Therapy. Front. Immunol. 2021, 12, 850. [Google Scholar] [CrossRef]
- Fucikova, J.; Palova-Jelinkova, L.; Bartunkova, J.; Spisek, R. Induction of Tolerance and Immunity by Dendritic Cells: Mechanisms and Clinical Applications. Front. Immunol. 2019, 10, 2393. [Google Scholar] [CrossRef]
- Suffner, J.; Hochweller, K.; Kühnle, M.-C.; Li, X.; Kroczek, R.A.; Garbi, N.; Hämmerling, G.J. Dendritic Cells Support Homeostatic Expansion of Foxp3+ Regulatory T Cells in Foxp3.LuciDTR Mice. J. Immunol. 2010, 1841, 1810. [Google Scholar] [CrossRef] [Green Version]
- Darrasse-Jèze, G.; Deroubaix, S.; Mouquet, H.; Victora, G.D.; Eisenreich, T.; Yao, K.; Masilamani, R.F.; Dustin, M.L.; Rudensky, A.; Liu, K.; et al. Feedback control of regulatory T cell homeostasis by dendritic cells in vivo. J. Exp. Med. 2009, 206, 1853–1862. [Google Scholar] [CrossRef] [Green Version]
- Hiasa, M.; Abe, M.; Nakano, A.; Oda, A.; Amou, H.; Kido, S.; Takeuchi, K.; Kagawa, K.; Yata, K.; Hashimoto, T.; et al. GM-CSF and IL-4 induce dendritic cell differentiation and disrupt osteoclastogenesis through M-CSF receptor shedding by up-regulation of TNF-α converting enzyme (TACE). Blood 2009, 114, 4517–4526. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, D.; Paul, K.; Bagchi, J.; Rakshit, S.; Mandal, L.; Bandyopadhyay, G.; Bandyopadhyay, S. Granulocyte-macrophage colony-stimulating factor drives monocytes to CD14low CD83+ DCSIGN- interleukin-10-producing myeloid cells with differential effects on T-cell subsets. Immunology 2007, 121, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Domogalla, M.P.; Rostan, P.V.; Raker, V.K.; Steinbrink, K. Tolerance through Education: How Tolerogenic Dendritic Cells Shape Immunity. Front. Immunol. 2017, 8, 1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansilla, M.J.; Contreras-Cardone, R.; Navarro-Barriuso, J.; Cools, N.; Berneman, Z.; Ramo-Tello, C.; Martínez-Cáceres, E.M. Cryopreserved vitamin D3-tolerogenic dendritic cells pulsed with autoantigens as a potential therapy for multiple sclerosis patients. J. Neuroinflamm. 2016, 13, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raϊch-Regué, D.; Grau-López, L.; Naranjo-Gómez, M.; Ramo-Tello, C.; Pujol-Borrell, R.; Martínez-Cáceres, E.; Borràs, F.E. Stable antigen-specific T-cell hyporesponsiveness induced by tolerogenic dendritic cells from multiple sclerosis patients. Eur. J. Immunol. 2012, 42, 771–782. [Google Scholar] [CrossRef]
- Mansilla, M.J.; González-Larreategui, I.; Figa-Martín, N.; Barallat, J.; Fondelli, F.; Sellés-Rius, A.; Quirant-Sánchez, B.; Teniente-Serra, A.; Martínez-Cáceres, E. Transfection of Vitamin D3-Induced Tolerogenic Dendritic Cells for the Silencing of Potential Tolerogenic Genes. Identification of CSF1R-CSF1 Signaling as a Glycolytic Regulator. Int. J. Mol. Sci. 2021, 22, 7363. [Google Scholar] [CrossRef]
- Marin, E.; Bouchet-Delbos, L.; Renoult, O.; Louvet, C.; Nerriere-Daguin, V.; Managh, A.J.; Even, A.; Giraud, M.; Vu Manh, T.P.; Aguesse, A.; et al. Human Tolerogenic Dendritic Cells Regulate Immune Responses through Lactate Synthesis. Cell Metab. 2019, 30, 1075–1090.e8. [Google Scholar] [CrossRef]
- Derdelinckx, J.; Mansilla, M.J.; De Laere, M.; Lee, W.-P.; Navarro-Barriuso, J.; Wens, I.; Nkansah, I.; Daans, J.; De Reu, H.; Jolanta Keliris, A.; et al. Clinical and immunological control of experimental autoimmune encephalomyelitis by tolerogenic dendritic cells loaded with MOG-encoding mRNA. J. Neuroinflamm. 2019, 16, 167. [Google Scholar] [CrossRef] [Green Version]
- Mansilla, M.J.; Sellès-Moreno, C.; Fàbregas-Puig, S.; Amoedo, J.; Navarro-Barriuso, J.; Teniente-Serra, A.; Grau-López, L.; Ramo-Tello, C.; Martínez-Cáceres, E.M. Beneficial Effect of Tolerogenic Dendritic Cells Pulsed with MOG Autoantigen in Experimental Autoimmune Encephalomyelitis. CNS Neurosci. Ther. 2015, 21, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Leng, X.; Li, H.; Yang, S.; Yang, T.; Li, L.; Xiong, Y.; Zou, Q.; Liu, Y.; Wang, Y. Tolerogenic Dendritic Cells Induced by BD750 Ameliorate Proinflammatory T cell Responses and Experimental Autoimmune Encephalitis in Mice. Mol. Med. 2017, 23, 204–214. [Google Scholar] [CrossRef]
- Mansilla, M.J.; Presas-Rodríguez, S.; Teniente-Serra, A.; González-Larreategui, I.; Quirant-Sánchez, B.; Fondelli, F.; Djedovic, N.; Iwaszkiewicz-Grześ, D.; Chwojnicki, K.; Miljković, Đ.; et al. Paving the way towards an effective treatment for multiple sclerosis: Advances in cell therapy. Cell. Mol. Immunol. 2021, 18, 1353–1374. [Google Scholar] [CrossRef]
- Willekens, B.; Presas-Rodríguez, S.; Mansilla, M.J.; Derdelinckx, J.; Lee, W.-P.; Nijs, G.; De Laere, M.; Wens, I.; Cras, P.; Parizel, P.; et al. Tolerogenic dendritic cell-based treatment for multiple sclerosis (MS): A harmonised study protocol for two phase I clinical trials comparing intradermal and intranodal cell administration. BMJ Open 2019, 9, e030309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zubizarreta, I.; Flórez-Grau, G.; Vila, G.; Cabezón, R.; España, C.; Andorra, M.; Saiz, A.; Llufriu, S.; Sepulveda, M.; Sola-Valls, N.; et al. Immune tolerance in multiple sclerosis and neuromyelitis optica with peptide-loaded tolerogenic dendritic cells in a phase 1b trial. Proc. Natl. Acad. Sci. USA 2019, 116, 8463–8470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strohl, W.R. Fusion Proteins for Half-Life Extension of Biologics as a Strategy to Make Biobetters. BioDrugs 2015, 29, 215–239. [Google Scholar] [CrossRef] [Green Version]
- Staykova, M.A.; Simmons, R.D.; Willenborg, D.O. Infusion of soluble myelin basic protein protects long term against induction of experimental autoimmune encephalomyelitis. Immunol. Cell Biol. 1997, 75, 54–64. [Google Scholar] [CrossRef]
- Devaux, B.; Enderlin, F.; Wallner, B.; Smilek, D.E. Induction of EAE in mice with recombinant human MOG, and treatment of EAE with a MOG peptide. J. Neuroimmunol. 1997, 75, 169–173. [Google Scholar] [CrossRef]
- Weiner, H.L.; Mackin, G.A.; Matsui, M.; Orav, E.J.; Khoury, S.J.; Dawson, D.M.; Hafler, D.A. Double-blind pilot trial of oral tolerization with myelin antigens in multiple sclerosis. Science 1993, 259, 1321–1324. [Google Scholar] [CrossRef]
- Freedman, M.S.; Bar-Or, A.; Oger, J.; Traboulsee, A.; Patry, D.; Young, C.; Olsson, T.; Li, D.; Hartung, H.-P.; Krantz, M.; et al. A phase III study evaluating the efficacy and safety of MBP8298 in secondary progressive MS. Neurology 2011, 77, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Chataway, J.; Martin, K.; Barrell, K.; Sharrack, B.; Stolt, P.; Wraith, D.C.; ATX-MS1467 Study Group. Effects of ATX-MS-1467 immunotherapy over 16 weeks in relapsing multiple sclerosis. Neurology 2018, 90, e955–e962. [Google Scholar] [CrossRef]
- Streeter, H.B.; Rigden, R.; Martin, K.F.; Scolding, N.J.; Wraith, D.C. Preclinical development and first-in-human study of ATX-MS-1467 for immunotherapy of MS. Neurol.-Neuroimmunol. Neuroinflamm. 2015, 2, e93. [Google Scholar] [CrossRef] [Green Version]
- Juryńczyk, M.; Walczak, A.; Jurewicz, A.; Jesionek-Kupnicka, D.; Szczepanik, M.; Selmaj, K. Immune regulation of multiple sclerosis by transdermally applied myelin peptides. Ann. Neurol. 2010, 68, 593–601. [Google Scholar] [CrossRef]
- Walczak, A.; Siger, M.; Ciach, A.; Szczepanik, M.; Selmaj, K. Transdermal Application of Myelin Peptides in Multiple Sclerosis Treatment. JAMA Neurol. 2013, 70, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Lutterotti, A.; Yousef, S.; Sputtek, A.; Stürner, K.H.; Stellmann, J.-P.; Breiden, P.; Reinhardt, S.; Schulze, C.; Bester, M.; Heesen, C.; et al. Antigen-Specific Tolerance by Autologous Myelin Peptide–Coupled Cells: A Phase 1 Trial in Multiple Sclerosis. Sci. Transl. Med. 2013, 5, 188ra75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kappos, L.; Comi, G.; Panitch, H.; Oger, J.; Antel, J.; Conlon, P.; Steinman, L.; Comi, G.; Kappos, L.; Oger, J.; et al. Induction of a non-encephalitogenic type 2 T helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase II trial. Nat. Med. 2000, 6, 1176–1182. [Google Scholar] [CrossRef] [PubMed]
- Bielekova, B.; Goodwin, B.; Richert, N.; Cortese, I.; Kondo, T.; Afshar, G.; Gran, B.; Eaton, J.; Antel, J.; Frank, J.A.; et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83–99) in multiple sclerosis: Results of a phase II clinical trial with an altered peptide ligand. Nat. Med. 2000, 6, 1167–1175. [Google Scholar] [CrossRef]
- Belogurov, A., Jr.; Zakharov, K.; Lomakin, Y.; Surkov, K.; Avtushenko, S.; Kruglyakov, P.; Smirnov, I.; Makshakov, G.; Lockshin, C.; Gregoriadis, G.; et al. CD206-Targeted Liposomal Myelin Basic Protein Peptides in Patients with Multiple Sclerosis Resistant to First-Line Disease-Modifying Therapies: A First-in-Human, Proof-of-Concept Dose-Escalation Study. Neurotherapeutics 2016, 13, 895–904. [Google Scholar] [CrossRef] [Green Version]
- Lomakin, Y.; Belogurov, A., Jr.; Glagoleva, I.; Stepanov, A.; Zakharov, K.; Okunola, J.; Smirnov, I.; Genkin, D.; Gabibov, A. Administration of Myelin Basic Protein Peptides Encapsulated in Mannosylated Liposomes Normalizes Level of Serum TNF-α and IL-2 and Chemoattractants CCL2 and CCL4 in Multiple Sclerosis Patients. Mediators Inflamm. 2016, 2016, 2847232. [Google Scholar] [CrossRef] [Green Version]
- Getts, D.R.; Martin, A.J.; McCarthy, D.P.; Terry, R.L.; Hunter, Z.N.; Yap, W.T.; Getts, M.T.; Pleiss, M.; Luo, X.; King, N.J.C.; et al. Microparticles bearing encephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis. Nat. Biotechnol. 2012, 30, 1217–1224. [Google Scholar] [CrossRef]
- Hunter, Z.; McCarthy, D.P.; Yap, W.T.; Harp, C.T.; Getts, D.R.; Shea, L.D.; Miller, S.D. A Biodegradable Nanoparticle Platform for the Induction of Antigen-Specific Immune Tolerance for Treatment of Autoimmune Disease. ACS Nano 2014, 8, 2148–2160. [Google Scholar] [CrossRef]
- Yeste, A.; Nadeau, M.; Burns, E.J.; Weiner, H.L.; Quintana, F.J. Nanoparticle-mediated codelivery of myelin antigen and a tolerogenic small molecule suppresses experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2012, 109, 11270–11275. [Google Scholar] [CrossRef] [Green Version]
- Hess, K.L.; Oh, E.; Tostanoski, L.H.; Andorko, J.I.; Susumu, K.; Deschamps, J.R.; Medintz, I.L.; Jewell, C.M. Engineering Immunological Tolerance Using Quantum Dots to Tune the Density of Self-Antigen Display. Adv. Funct. Mater. 2017, 27, 1700290. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.J.; Stewart, J.M.; Drashansky, T.T.; Brusko, M.A.; Zuniga, A.N.; Lorentsen, K.J.; Keselowsky, B.G.; Avram, D. An antigen-specific semi-therapeutic treatment with local delivery of tolerogenic factors through a dual-sized microparticle system blocks experimental autoimmune encephalomyelitis. Biomaterials 2017, 143, 79–92. [Google Scholar] [CrossRef]
- Saito, E.; Kuo, R.; Kramer, K.R.; Gohel, N.; Giles, D.A.; Moore, B.B.; Miller, S.D.; Shea, L.D. Design of biodegradable nanoparticles to modulate phenotypes of antigen-presenting cells for antigen-specific treatment of autoimmune disease. Biomaterials 2019, 222, 119432. [Google Scholar] [CrossRef]
- Cappellano, G.; Woldetsadik, A.D.; Orilieri, E.; Shivakumar, Y.; Rizzi, M.; Carniato, F.; Gigliotti, C.L.; Boggio, E.; Clemente, N.; Comi, C.; et al. Subcutaneous inverse vaccination with PLGA particles loaded with a MOG peptide and IL-10 decreases the severity of experimental autoimmune encephalomyelitis. Vaccine 2014, 32, 5681–5689. [Google Scholar] [CrossRef]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Ernsting, M.J.; Murakami, M.; Roy, A.; Li, S.-D. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J. Control. Release 2013, 172, 782–794. [Google Scholar] [CrossRef] [Green Version]
- Getts, D.R.; Shea, L.D.; Miller, S.D.; King, N.J.C. Harnessing nanoparticles for immune modulation. Trends Immunol. 2015, 36, 419–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maldonado, R.A.; LaMothe, R.A.; Ferrari, J.D.; Zhang, A.-H.; Rossi, R.J.; Kolte, P.N.; Griset, A.P.; O’Neil, C.; Altreuter, D.H.; Browning, E.; et al. Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance. Proc. Natl. Acad. Sci. USA 2015, 112, E156–E165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
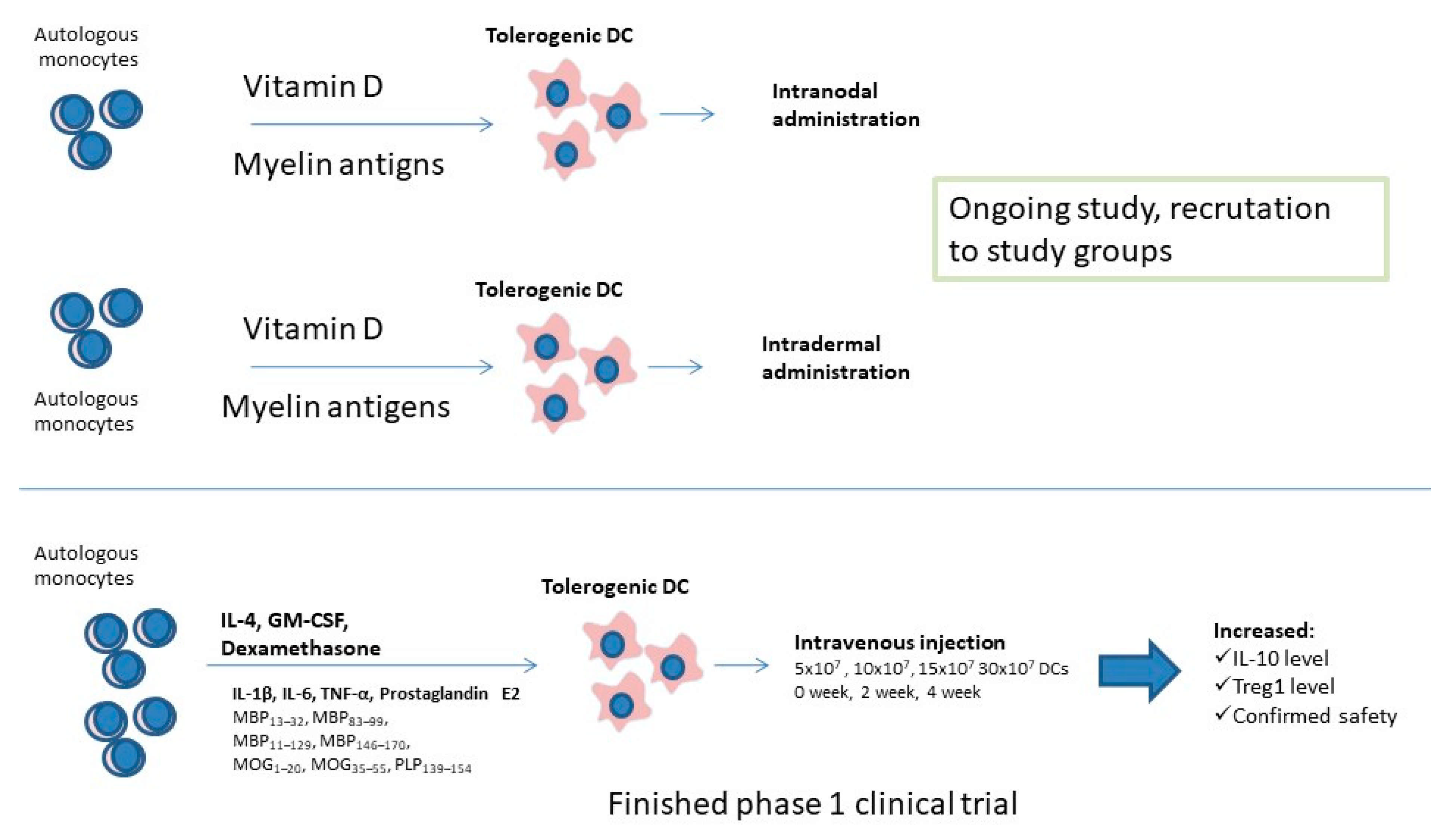
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).