Abstract
Gains and losses of large segments of genomic DNA, known as copy number variants (CNVs) gained considerable interest in clinical diagnostics lately, as particular forms may lead to inherited genetic diseases. In recent decades, researchers developed a wide variety of cytogenetic and molecular methods with different detection capabilities to detect clinically relevant CNVs. In this review, we summarize methodological progress from conventional approaches to current state of the art techniques capable of detecting CNVs from a few bases up to several megabases. Although the recent rapid progress of sequencing methods has enabled precise detection of CNVs, determining their functional effect on cellular and whole-body physiology remains a challenge. Here, we provide a comprehensive list of databases and bioinformatics tools that may serve as useful assets for researchers, laboratory diagnosticians, and clinical geneticists facing the challenge of CNV detection and interpretation.
1. Introduction
Among the least understood types of genetic variation are copy number variants (CNVs), a class of unbalanced structural variants characterized by deletions, insertions, duplications or even multiplications of DNA segments ranging in size from a few dozen of bp up to several Mb. Currently, the lower limit for CNV length is 50 bps, but this value has been gradually decreasing due to continuous methodological progress. The shift is mainly due to an increased resolution of used methods, allowing for detection of a wider variety of variant lengths and for an increase of CNV detection capacity (Figure 1). Considering this remarkable shift and the fact that generally used distinguishing criteria are somewhat vaguely defined, it has also been suggested that CNVs include a wider spectrum of variants. However, for practical reasons, we will focus on the conventional concept of CNVs in this review.
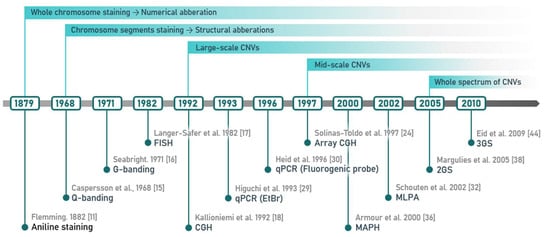
Figure 1.
Hallmarks in copy number variant (CNV) history. The 20th century saw a steady development of methods, which finally allowed genome-wide, high-resolution CNV detection around the beginning of the 21st century.
Pushing the limits of CNV detection revealed that they are widespread in human populations with a 5–10% difference of genomic sequences between normal individuals [1,2,3]. As a significant aspect of our heterogeneity, CNVs may disrupt gene function or alter gene dosage by direct gain or loss of coding sequences [4], but several indirect mechanisms including alteration of non-coding RNAs [5,6] and topologically associated domains [7] have been described. Since these may affect the phenotype, CNVs may threaten the ability to survive or, on the contrary, enhance chances of survival in disadvantageous environments [8].
CNVs are an important cause of genomic disorders with Mendelian inheritance, and may also contribute to complex diseases with multifactorial etiology [9]. Since the introduction of high throughput technologies for CNV detection, namely array-based comparative genomic hybridization (aCGH) and massively parallel sequencing (MPS), the number of novel variants is constantly increasing. However, a lot of detected CNVs are still categorized as variants of uncertain significance (VUS) with unknown clinical impact [4], suggesting a need for their reliable classification. Therefore, all available information has been translated to the standards of interpretation and reporting of constitutional CNVs as recently published by the American College of Medical Genetics and Genomics and the Clinical Genome Resource [10]. However, the age of high throughput technologies comes with an ever increasing amount of generated data. Researchers must continuously improve bioinformatic softwares and decision support tools to help clinicians handle the problem.
2. Methods of CNV Detection
From conventional cytogenetic methods through hybridization- and PCR-based techniques, up to MPS, CNV detection methods have been through a long evolution, affecting several aspects of progress in CNV research (Figure 1). Cytogenetic techniques were the first methods for CNV detection, based on visual inspection of chromosomes. Improvements led to the gradual lowering of detection limits, from numerical anomalies of whole chromosomes to CNVs of a few Mb in size. Introduction of molecular-biology methods, especially hybridization followed by Southern-blotting, allowed detection of mid-sized CNVs in the range of several kb. Later, amplification-based PCR methods together with their modifications and a wide range of associated detection techniques brought analytical resolution to single nucleotides, with upper limits of the detection range at hundreds of kb or a few Mb. Completely new possibilities were introduced by housing molecular hybridization techniques with cytogenetic methods and with microarray-based methods, but also with the invention of MPS. The latter two allowed the analysis of the whole size range of CNVs in single runs, at least theoretically, and in scales of whole genomes.
2.1. Cytogenetic Techniques and Their Most Common Modifications
Although chromosomes in plant and animal cells were first observed in the 19th century [11], and CNVs were microscopically detected in Drosophila in the early 20th century [12], it took the first half of the 20th century to assess the human diploid karyotype [13]. This was finally allowed by several methodological improvements in karyotyping, leading to an establishment of conventional cytogenetic techniques which are still in general use. These include the use of cells cultured from the tested tissue, arresting of dividing cells in metaphase by colchicine, treatment by a hypotonic solution to spread the chromosomes, fixation of chromosomes on a glass slide for examination under a light microscope, and subsequent counting and grouping of chromosomes according to their morphological features [14]. A revolutionary step in human cytogenetics came with the introduction of different chromosome banding techniques revealing specific chromosomal patterns, including fluorescence-based Quinacrine banding (Q-banding) [15] and Giemsa staining (G-banding) [16], which have become the most widely used banding methods. Following the advent of cytogenetic and banding techniques, discoveries were quickly made with regard to CNVs associated with human pathologies. However, karyotyping techniques available in the 1960s only allowed detection of gross numerical and morphological abnormalities, because the resolution of light microscopes was limited to imbalances larger than 5 Mb.
Conventional cytogenetic techniques combined with molecular techniques such as hybridisation led to the emergence of molecular cytogenetics, the main methods being fluorescence in situ hybridization (FISH) [17] and comparative genomic hybridization (CGH), both still requiring fluorescent microscopy [18]. FISH is based on hybridisation of sequence specific fluorescently labelled probes with subsequent microscopic detection of a given fluorescent signal that indicates the presence or absence of specific target DNA sequences [17]. This technique has undergone several modifications, from single-event-specific tests up to chromosome painting, making it possible to detect individual loci as small as 10 kb [19]. Certain limitations of conventional cytogenetics and FISH, mainly those of resolution, led to the development of CGH for CNV detection [18]. By the comparison of fluorescent signals generated from DNA of the tested and control samples, along the chromosomes to which they were hybridised, CGH is capable of identifying increased or decreased copy numbers of sequences at least ~3–10 Mb in size [20].
2.2. Methods of Molecular Biology
Molecular-biology methods such as Southern blot hybridization offer higher resolution than cytogenetics [21]. The principle of Southern blotting relies upon fragmentation of DNA with a restriction endonuclease and separating fragments by gel electrophoresis. The fragments are transferred to a membrane and hybridized to appropriate probes. Copy number changes are visible as differential hybridization intensities or as altered mobility of the fragments. Although for many years, Southern blotting was the standard method for the detection of deletions or amplifications in the range of 5–500 kb [22], it is a laborious, time-consuming method that requires large amounts of high-quality DNA [23].
A tremendous improvement in the screening of CNVs came with the introduction of microarray-based methods, specifically in conjunction with comparative genomic hybridization, where DNA samples extracted from the tested and reference cells are cohybridized to an array of fixed oligonucleotide probes instead of metaphase chromosomes [24]. The aCGH provides genome-wide coverage at a much higher resolution of 10–25 kb [25] or even >500 bp if high-density arrays are used [26]. Despite some limitations in resolution and accuracy, this made aCGH a standard in CNV detection [27].
PCR-based methods may either come in the form of conventional two- or three-primer based protocols or multiplexed assays. CNVs may be detected by PCR-based protocols through the change of: (i) migrational properties during agarose gel or other types of electrophoresis [28]; (ii) amplification cycles required to achieve a relative threshold fluorescent intensity in real-time quantitative PCR (qPCR) assays [29,30]; (iii) relative fluorescent signal intensities in capillary electrophoresis when using quantitative-fluorescent PCR (QF-PCR) [31] or multiplex ligation-dependent probe amplification (MLPA) techniques [32]; (iv) denaturation properties reflected in melting temperatures or melting curve shapes during conventional or high-resolution melting analysis [33]. All of these methods are more or less convenient for the targeted detection of a limited number of CNVs in a relatively wide range of length from tens of bp up to Mb and even whole chromosomes at low cost and fast turnaround time [34]. However, each of these methods also has its own advantages and limitations. A more recently introduced alternative to the traditional qPCR in CNV detection is the droplet digital PCR (ddPCR). In this method, template DNA is diluted and partitioned into thousands of nano-scale droplets of uniform volume, allowing for absolute quantification of target copy numbers without the need for a standard assay, making results easier to interpret and less error-prone than regular qPCR [35]. Another PCR-based method is multiplex amplifiable probe hybridization (MAPH) using oligonucleotide probes that hybridize to a specific region in the genome. Hybridized probes are amplified and the amount of each amplification product is proportional to the copy number of the corresponding sequence [36]. MAPH enables the sensitive detection of CNVs as small as 150 bp [37]. Even more sensitive and easier to use is MLPA developed to determine the copy number of multiple genomic DNA sequences (up to 60 probes) in a single reaction with resolution from a single nucleotide difference. The probes hybridized to the sample DNA are ligated and amplified, resulting in fragments of a unique length which can be separated and quantified by capillary electrophoresis [32]. Therefore, MLPA is a cost-effective method that can be performed with equipment present in most molecular biology laboratories.
Although the first generation of DNA sequencing (1GS) technologies, specifically Sanger sequencing, was generally considered to be the gold standard in DNA diagnostics, CNVs represent specific challenges not easily dealt with when using this method. Their detectability strongly depends on the length and type of the CNV, as well as on its position with regard to the used amplification primers (for more details on the possible effect of CNVs on reliability, see the next subheading). Since 2005, when the first platforms of second-generation sequencing (2GS) technology became available [38], methods based on MPS have undergone several modifications, their cost ever on the decrease [39]. In current times, 2GS represents a valuable tool for clinical diagnostics and provides a sensitive and accurate approach for the detection of the major types of genomic variations, including CNVs [40]. There are three main strategies for 2GS-based CNV analysis, namely whole-genome, whole-exome, and targeted sequencing. Due to the limited length of DNA fragments sequenced by 2GS sequencers, variation is detected by abnormalities in the affected areas using robust statistical and bioinformatic processing [41]. Read-depth methods highlight regions with an irregular number of sequenced fragments: a loss is seen as a lower, and a gain as a higher than expected amount of a particular segment. Read-pair and split-read approaches analyze fragments with discordant alignments of sequenced fragments, where portions of a single fragment are aligned to unexpected sites in the reference genome. While the above methods directly analyze reads mapped to the reference genome, assembly-based methods compare longer sections of an individual’s genome, called contigs. This approach may reveal more complex genome rearrangements, but genome assembly is computationally more intensive and requires substantially higher capacity. Whole-genome sequencing combined with sophisticated computational strategies improved CNV detection, allowing even base-pair resolution of breakpoints [42]. On the other hand, whole-exome sequencing targets only the protein-coding part of the genome. However, since most of the known disease-causing mutations fall into this category, exome sequencing significantly reduces sequencing cost in medical applications and is still sufficiently powerful. Moreover, targeted sequencing provides a greater depth of coverage in regions of interest for an even lower cost [43].
Third-generation sequencing (3GS) technologies (e.g., single-molecule real-time sequencing [44] and nanopore sequencing [45]) bring promise for better characterization of genomic structural variants due to longer reads [46] that can be more confidently aligned to repetitive sequences, often mediating the formation of structural variants [47]. While both microarray and 2GS techniques are based on complex laboratory procedures which require several days to obtain results [48], nanopore-based 3GS provides pocket-sized, low-cost devices that usually take from 24 to 48 h to run, with reads generated continuously, so data can be used for processing and further analysis in real-time during the ongoing sequencing process [45]. Moreover, the method can be combined with a rapid library preparation kit capable of obtaining ready to sequence genomic DNA in 10 min. Data generated in the first tens of minutes of a run are sufficient to detect large chromosomal alterations with a resolution in the order of tens of Mb. Data produced in the first 6–12 h of a sequencing run can be used to identify CNVs with an accuracy comparable to currently available array-based methods, and are capable of predicting the allelic fraction of genomic alterations with high accuracy [42]. The problem with CNV breakpoint identification often encountered in PCR- or array-based methods can also be solved by breakpoint sequencing [49]. Using 3GS devices, it will soon be possible to perform a cost-effective high-resolution molecular karyotyping of the human genome within an hour from sample extraction, allowing ultra-fast analyses in fields where time matters, such as precision oncology and prenatal diagnostics [42].
When considering in silico tools to extract CNV genotype information from generated data, nearly all of the available methods have their dedicated commercial tools, from cytogenetic karyotyping, through MLPA, up to aCGH. The bioinformatic tools for processing MPS data are, however, still under intensive development and diversification (Table 1). While both 2GS and 3GS are technically capable of detecting CNVs in a wide range of length, not each size is identifiable using the same bioinformatics pipeline and different variants may require differently suited tools [50]. To identify smaller structural variations spanning several nucleotides, conventional variant callers, such as the GATK HaplotypeCaller [51], are generally suitable, while large CNVs exceeding read lengths are typically identified based on a disproportion of sequenced reads from the genomic region of a particular CNV [52]. In conclusion, since each CNV detecting method has its advantages and limitations, the choice for an appropriate technique depends on the application, required resolution, available lab equipment, workload, and budget.

Table 1.
Bioinformatic tools for detection of CNVs from next generation sequencing-based genomic data. Several tools are capable of detection and annotation of CNVs at the same time (e.g., iCopyDAV, SG-ADVISER-CNV, DeAnnCNV), so they are listed in the next table. WES (whole-exome sequencing); WGS (whole-genome sequencing).
2.3. Techniques Possibly Affected by the Presence of Undetected CNVs
In addition to detection possibilities, another aspect worth discussing is that certain methods are at risk of giving inferior results due to the presence of undetected CNVs. Such methods include Southern blotting and PCR as well as both 1GS and 2GS. PCR and PCR-based sequencing methods are prone to allelic dropout caused by the presence of deletions in the analyzed region, especially if affecting one of the primer binding regions, or may falsely show hemizygous instead of homozygous alleles if the entire amplified region is deleted. They may also be affected by the presence of false-positive variants, such as unknown homologous copies of the analyzed region (e.g., pseudogenes or pseudoexons) with high but not full sequence homology, like in the case of the CFTR pseudoexon 2 present in the GRCh38, but not in earlier versions of the human reference genome [50].
Some of these effects may be prevented, eliminated, or at least attenuated in some ways, depending on whether the presence of a certain CNV is expected or unforeseen. These methods include but are not limited to: (i) checking the region of interest for specific CNVs by an alternative technique (e.g., sequencing of single genes may be complemented by MLPA, while sequencing of whole exomes and genomes may incorporate a CNV-specific bioinformatic variant calling pipeline to complement conventional variant calling of small variants); (ii) using two or more complementing assays based on different principles and being liable to different biases; (iii) careful evaluation and reporting of results by well trained users who are familiar with the used technique, including thorough quality control and reporting only unambiguous findings truly supported by the results ( for example, not reporting variants as homozygous, when detected using sequencing with PCR preamplification, unless other heterozygous variants in the same amplicon were not detected, or until the possible presence of CNVs is checked); or (iv) at least by disclosing the possible biases in the results.
3. Potential Biomedical Applications of CNV Detection
CNVs can be analyzed from different biological sources, offering various valuable information, so there are plenty of biomedical applications where CNV detection may be useful. CNVs have been studied in neuropsychiatric [72,73], developmental [74], and cardiovascular diseases [75]. Several studies have identified the role of CNVs in common diseases such as coronary artery disease or in rarer events such as sudden cardiac death. Such findings may be useful for clinicians for disease classification and detection in the future, particularly in the age of the whole genome sequencing [76].
On the other hand, CNVs have been identified as susceptibility factors for autoimmune diseases such as systemic lupus erythematosus (SLE). The human C4 gene is one of the most striking examples of genetic diversity, due to a great variation in number and size of gene copies between individuals. Low copy numbers of the C4 and C4A gene are significant risk factors for the development of SLE in different populations. Meta-analysis by Li et al. showed that <4 copies of the C4 gene increase susceptibility to autoimmune diseases with an odds ratio of 1.46 (95% CI, 1.19–1.78) [77]. In addition, C4A has been associated with disease severity. Thus, determination of C4 gene copy numbers may be useful in sub-phenotyping and managing SLE patients [78].
CNVs obtained from blood cells or tissues are suitable for the identification of germline or somatic variants. Tissue biopsy is a well-established procedure in cancer diagnosis for identification of human genomic alterations. However, this technique is invasive, time-consuming, not sufficient to examine the entire tumor profile, and not applicable in the follow-up of cancer treatment [79]. The current trend is moving towards non- or less-invasive sampling, such as liquid biopsy [80]. In combination with whole-genome copy number analysis, which does not require any prior knowledge about the characteristics of the primary tumor genome, it represents a promising clinical tool. Heitzer et al. reviewed approaches for analyses of somatic copy number alterations at a genome-wide scale [81]. Both circulating tumor cells (CTCs) and cell-free DNA (cfDNA) were shown to be powerful sources in CNV profiling.
Ni et al. hypothesize that copy number changes are key events of metastasis. They observed cancer-associated CNVs in exomes of CTCs revealing information needed for individualized therapy, such as drug resistance and phenotypic transition and suggest that CNVs at certain genomic loci have the potential for CTC-based cancer diagnostics [82]. Several studies demonstrated that the detection of ALK gene rearrangement in non–small-cell lung cancer (NSCLC), a predictive biomarker for crizotinib treatment, may be performed using CTCs. The same group also reported that CTCs can be used for sensitive detection of ROS1 rearrangement in NSCLC patients. CTCs from ROS1-rearranged patients show heterogeneity of ROS1 gene abnormalities and elevated numerical chromosomal instability, suggesting a potential mechanism for resistance to crizotinib, a known ROS1-inhibitor [83].
Since tumor cells frequently undergo necrosis, they release tumor-specific cfDNA (ctDNA) into body fluids such as blood, urine, saliva, etc. [84]. It was shown that quantification of tumor-specific rearrangements in ctDNA by ddPCR is highly accurate for postsurgical discrimination between patients with an eventual diagnosis of clinical metastasis and long-term disease-free patients, with a sensitivity of 93% (95% CI, 66–100%) and specificity of 100% (95% CI, 61–100%). Moreover, ctDNA-based detection preceded clinical detection of metastasis in 86% of patients with an average lead time of 11 months, whereas patients with long-term disease-free survival had undetectable ctDNA postoperatively [85]. Peng et al. presented a method enabling CNV detection from a 150-gene panel using a low amount of ctDNA. They demonstrated that their CNV pipeline can detect EGFR, ERBB2, and MET amplification from ctDNA samples with high specificity and concordance with corresponding tissue-based whole-exome results. The concordance rate for EGFR, ERBB2, and MET CNVs was 78%, 89.6%, and 92.4%, respectively [86]. The analysis of circulating nucleic acids may also be helpful in other diseases. Since cfDNA biomarkers are known to be important in many autoimmune and multifactorial diseases such as IBD [87], cfDNA could also be used for studying CNVs in such disorders.
CNVs are also useful in the diagnostics of rare and common diseases or predispositions. This may be performed as prenatal testing through direct testing of the fetus or indirectly using maternal blood. Detection of CNVs is a common part of modern non-invasive prenatal testing (NIPT), most commonly based on low-coverage whole-genome sequencing analysis of cell-free fetal DNA (cffDNA) from maternal plasma [88]. This approach is useful for the detection of chromosomal aneuploidy and microdeletion syndromes, including DiGeorge, Prader-Willi/Angelman, 1p36, Cri-du-chat, and Wolf-Hirschhorn syndrome [52]. Apart from fetal CNVs, maternal ones can also be detected by this method, although current analyses generally do not interpret these findings. Maternal aberrations are potentially harmful to the fetus, so some authors suggest reporting these variants if clinically relevant. On the other hand, performing NIPT may lead to the incidental diagnosis of maternal diseases, such as previously unrecognized pathologies, late-onset diseases and predispositions arising from maternal germline CNVs, or malignancies and systemic autoimmune diseases presenting with somatic CNVs. Thus, these aspects of CNV detection also affect conventional perception of incidental and secondary findings arising via genetic testing, which are now extensively discussed [89]. Giles et al. reported that 80% of genetic counselors recognized it would be beneficial to use NIPT for neoplasm screening, yet more than 90% affirmed that guidelines are necessary to prepare for such situations [90].
CNV detection may also find application in the evaluation of the microbiome balance, through the analysis of CNVs in metagenomes in different body parts. The human microbiome interacts with the host and plays an important role in many host biological processes [91]. Host genomic variations influence the composition of the microbiome, which in turn affects the health of the individual. While numerous studies have been focused on associations between the gut microbiome and specific alleles of the host genome, gene copy number also varies. It was shown, for instance, that duplication of the human AMY1 gene is associated with an increased number of oral Porphyromonas in saliva, which is linked to periodontitis. Gut microbiota of these individuals had increased abundance of resistant starch-degrading microbes, produced higher levels of short-chain fatty acids, and drove higher adiposity when transferred to germ-free mice [92]. This case demonstrated that even seemingly harmless variants in the host genome could affect the health of an individual.
Current knowledge suggests that it is important to analyze CNVs not only in human cells, but also in the microbiome. Taxonomic characterization of the human microbiota is often limited to the species level, however, each microbial species represents a large collection of strains that may contain considerably different sets or copy numbers of genes resulting in potentially distinct functional capacities. This intra-species variation is caused by deletion and duplication events, which were shown to be prevalent in the human gut environment, with some species exhibiting CNVs in >20% of their genes. This variability is especially prevalent in disease-associated genes involved in important functions, such as transport and signaling. A study by Greenblum et al. showed obesity to be associated with higher copy numbers of thioredoxin 1 in Clostridium sp., an increased copy number of an MFS transporter gene in the Roseburia inulinivorans genome cluster, and increased HlyD in Bacteroides uniformis associated with IBD-afflicted individuals. According to the authors, the analysis of species composition alone is not sufficient to capture the true functional potential of the microbiome because it may fail to capture important functional differences, so the analysis of intra-species variation in microbial communities is crucial [93].
4. Clinical Interpretation of CNVs
As detailed above, CNVs are an important source of normal and pathogenic variation. Pathogenic CNVs are typically large and contain multiple genes, significantly enriched in developmental genes and genes with greater evolutionary copy number conservation across mammals. On the other hand, genes found in benign CNVs have more variable copy numbers, suggesting that dosage sensitivity of genes is a predominant causative factor for CNV pathogenicity [94]. In everyday practice, laboratory diagnosticians, genetic counselors, and clinical geneticists need to distinguish pathogenic CNVs from benign ones in their patients, and such interpretation can be challenging. Many recurring CNVs are already classified into one of the five main classes of clinical impact (benign, likely benign, VUS, likely pathogenic, and pathogenic), a uniformized system commonly used for the interpretation of other sequence variants as well [95]. However, progress in the detection of CNVs resulted in a growing amount of novel CNVs that need further analysis to determine their potential clinical impact, while between the two clear extremes (benign and pathogenic), a wide spectrum of CNVs lacking evidence to support their clinical significance are classified as VUS [4]. This led to a demand for a more convenient annotation and classification of such CNVs. Even though the prediction of the clinical impact of CNVs is a challenge, there are several in silico prediction or decision support tools (Table 2) for CNV classification to help laboratory diagnosticians, genetic counselors and clinicians [96].
Table 2.
Decision support tools for annotation and/or classification of CNVs. SV (structural variation); WGS (whole-genome sequencing); WES (whole-exome sequencing); TADs (topologically associated domains).
It is essential to produce consistent, evidence-based clinical classification across laboratories and accurate clinical interpretation of CNVs, which requires not only appropriate methods to evaluate genomic content but also correlating clinical findings with reports in the medical literature. To ensure this, existing standards for evaluating CNVs were recently updated, and detailed recommendations for the interpretation and reporting of constitutional CNVs were published [10]. These recommendations comprise a semiquantitative point-based scoring system in which evidence categories with assigned relative weight were determined. When evaluating individual CNVs, genomic content, dosage sensitivity, predicted functional effect, clinical overlap with patients in the literature, evidence from case and control databases (Table 3), and de novo occurrence or inheritance patterns are considered [10]. Using this scoring system, any evaluated CNV should be assigned to one of the five above mentioned main classes of clinical impact [95]. It was also demonstrated that topologically associated domains, in which structural alteration results in various malformations, may increase clinical suspicion of pathogenicity for variants of uncertain significance. This piece of information, among others, may help in the clinical interpretation of CNVs that would otherwise be ignored based on current reporting criteria [106]. So, appropriate clinical interpretation relies on supporting evidence and, therefore, is still challenging. An effective way of overcoming the problem of VUS and achieving progress in clinical interpretation that may eventually translate to an improvement in patient health care is to share data and relevant information between laboratories and researchers [110].
Table 3.
Databases of common and clinically relevant genomic CNVs. The most popular databases that play a crucial role in variant classification are listed here.
5. Conclusions
In this work, we provide an overview of CNV detection methods, from basic cytogenetic methods to molecular-based approaches such as aCGH or MPS. Detecting CNVs in individuals and within populations is essential to better understand our genome and to elucidate its possible contribution to disease or phenotype. The growing availability of sequencing technology can help to further explore these functional implications, but since it can yield up to several terabytes of genomic data per run, it is not possible to unlock the full potential of such data without the help of CNV-related bioinformatic tools. Despite all the improvements in methodology and software, clinical interpretation of CNVs still remains a major challenge. Moreover, due to improving resolution, the number of novel structural variants is constantly increasing and this led to a demand for more convenient tools designed for storing, searching, annotating and evaluating CNV-related data to increase practical value for researchers, laboratory diagnosticians and clinical geneticists facing the challenging task of correctly interpreting the clinical impact of CNVs.
Author Contributions
Conceptualization, O.P. and J.R.; investigation, O.P., G.B. and Z.P.; writing—original draft preparation, O.P., J.R., J.S., Z.P. and G.B.; writing—review and editing, J.R., G.B., J.B. and B.N.; visualization, O.P.; supervision, O.P. and T.S.; funding acquisition, M.K. and T.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the OP Integrated Infrastructure for the project: Center for biomedical research [ITMS: 313011W428]; the OP Research and Development for the project: Research centre for severe diseases and related complications [ITMS: 26240120038]; and was also supported by the OP Integrated Infrastructure for the project: Long term strategic research and development focused on the occurrence of Lynch syndrome in the Slovak population and possibilities of prevention of tumors associated with this syndrome [ITMS: 313011V578] each co-financed by the European Regional Development Fund.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
O.P., J.R., Z.P., J.B., and T.S. are employees of Geneton Ltd., which participated in the development of commercial NIPT and GenomeScreen tests for the detection of CNVs. All remaining authors, namely J.S., G.B., M.K., and N.B. declare no potential conflict of interest.
References
- Sebat, J.; Lakshmi, B.; Troge, J.; Alexander, J.; Young, J.; Lundin, P.; Månér, S.; Massa, H.; Walker, M.; Chi, M.; et al. Large-scale copy number polymorphism in the human genome. Science 2004, 305, 525–528. [Google Scholar] [CrossRef]
- Iafrate, A.J.; Feuk, L.; Rivera, M.N.; Listewnik, M.L.; Donahoe, P.K.; Qi, Y.; Scherer, S.W.; Lee, C. Detection of large-scale variation in the human genome. Nat. Genet. 2004, 36, 949–951. [Google Scholar] [CrossRef] [PubMed]
- Zarrei, M.; MacDonald, J.R.; Merico, D.; Scherer, S.W. A copy number variation map of the human genome. Nat. Rev. Genet. 2015, 16, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Nowakowska, B. Clinical interpretation of copy number variants in the human genome. J. Appl. Genet. 2017, 58, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Gu, X.; Wang, G.; Huang, Y.; Ju, S.; Huang, J.; Wang, X. Copy number variations primed lncRNAs deregulation contribute to poor prognosis in colorectal cancer. Aging 2019, 11, 6089–6108. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, M.; Krishnan, P.; Cass, C.E.; Hubaux, R.; Lam, W.; Yasui, Y.; Damaraju, S. Breast cancer associated germline structural variants harboring small noncoding RNAs impact post-transcriptional gene regulation. Sci. Rep. 2018, 8, 7529. [Google Scholar] [CrossRef]
- Lupiáñez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef]
- Bennett, P.M. Genome plasticity: Insertion sequence elements, transposons and integrons, and DNA rearrangement. Methods Mol. Biol. 2004, 266, 71–113. [Google Scholar] [CrossRef]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef]
- Flemming, W. Zellsubstanz, Kern und Zelltheilung; FCW Vogel: Leipzig, Germany, 1882. [Google Scholar]
- Tice, S.C. A new sex-linked character in drosophila. Biol. Bull. 1914, 26, 221–230. [Google Scholar] [CrossRef]
- Tjio, J.H.; Levan, A. The chromosome number of man. Hereditas 2010, 42, 1–6. [Google Scholar] [CrossRef]
- Moorhead, P.S.; Nowell, P.C.; Mellman, W.J.; Battips, D.M.; Hungerford, D.A. Chromosome preparations of leukocytes cultured from human peripheral blood. Exp. Cell Res. 1960, 20, 613–616. [Google Scholar] [CrossRef]
- Caspersson, T.; Farber, S.; Foley, G.E.; Kudynowski, J.; Modest, E.J.; Simonsson, E.; Wagh, U.; Zech, L. Chemical differentiation along metaphase chromosomes. Exp. Cell Res. 1968, 49, 219–222. [Google Scholar] [CrossRef]
- Seabright, M. A rapid banding technique for human chromosomes. Lancet 1971, 2, 971–972. [Google Scholar] [CrossRef]
- Langer-Safer, P.R.; Levine, M.; Ward, D.C. Immunological method for mapping genes on Drosophila polytene chromosomes. Proc. Natl. Acad. Sci. USA 1982, 79, 4381–4385. [Google Scholar] [CrossRef]
- Kallioniemi, A.; Kallioniemi, O.P.; Sudar, D.; Rutovitz, D.; Gray, J.W.; Waldman, F.; Pinkel, D. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 1992, 258, 818–821. [Google Scholar] [CrossRef]
- Trask, B.J. Human cytogenetics: 46 chromosomes, 46 years and counting. Nat. Rev. Genet. 2002, 3, 769–778. [Google Scholar] [CrossRef]
- Bejjani, B.A.; Saleki, R.; Ballif, B.C.; Rorem, E.A.; Sundin, K.; Theisen, A.; Kashork, C.D.; Shaffer, L.G. Use of targeted array-based CGH for the clinical diagnosis of chromosomal imbalance: Is less more? Am. J. Med. Genet. 2005, 134, 259–267. [Google Scholar] [CrossRef]
- Southern, E.M. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J. Mol. Biol. 1975, 98, 503–517. [Google Scholar] [CrossRef]
- Lee, J.H.; Jeon, J.T. Methods to detect and analyze copy number variations at the genome-wide and locus-specific levels. Cytogenet. Genome Res. 2008, 123, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Hoebeeck, J.; Speleman, F.; Vandesompele, J. Real-Time Quantitative PCR as an Alternative to Southern Blot or Fluorescence in situ Hybridization for Detection of Gene Copy Number Changes. In Protocols for Nucleic Acid Analysis by Nonradioactive Probes; Humana Press: Totowa, NJ, USA, 2007; pp. 205–226. [Google Scholar]
- Solinas-Toldo, S.; Lampel, S.; Stilgenbauer, S.; Nickolenko, J.; Benner, A.; Döhner, H.; Cremer, T.; Lichter, P. Matrix-based Comparative Genomic Hybridization: Biochips to Screen for Genomic Imbalances. Genes Chromosomes Cancer 1997, 20, 399–407. [Google Scholar] [CrossRef]
- Yoon, S.; Xuan, Z.; Makarov, V.; Ye, K.; Sebat, J. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 2009, 19, 1586–1592. [Google Scholar] [CrossRef] [PubMed]
- Alkan, C.; Coe, B.P.; Eichler, E.E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 2011, 12, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, C.R., 2nd; Scharer, G.H.; Shaikh, T.H. Clinical impact of copy number variation analysis using high-resolution microarray technologies: Advantages, limitations and concerns. Genome Med. 2012, 4, 80. [Google Scholar] [CrossRef]
- Mahadevan, M.S.; Foitzik, M.A.; Surh, L.C.; Korneluk, R.G. Characterization and polymerase chain reaction (PCR) detection of an Alu deletion polymorphism in total linkage disequilibrium with myotonic dystrophy. Genomics 1993, 15, 446–448. [Google Scholar] [CrossRef]
- Higuchi, R.; Fockler, C.; Dollinger, G.; Watson, R. Kinetic PCR analysis: Real-time monitoring of DNA amplification reactions. Biotechnology 1993, 11, 1026–1030. [Google Scholar] [CrossRef]
- Heid, C.A.; Stevens, J.; Livak, K.J.; Williams, P.M. Real time quantitative PCR. Genome Res. 1996, 6, 986–994. [Google Scholar] [CrossRef]
- Yau, S.C.; Bobrow, M.; Mathew, C.G.; Abbs, J.S. Accurate diagnosis of carriers of deletions and duplications in Duchenne/Becker muscular dystrophy by fluorescent dosage analysis. J. Med. Genet. 1996, 33, 550–558. [Google Scholar] [CrossRef][Green Version]
- Schouten, J.P.; McElgunn, C.J.; Waaijer, R.; Zwijnenburg, D.; Diepvens, F.; Pals, G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002, 30, e57. [Google Scholar] [CrossRef]
- Radvansky, J.; Resko, P.; Surovy, M.; Minarik, G.; Ficek, A.; Kadasi, L. High-resolution melting analysis for genotyping of the myotonic dystrophy type 1 associated Alu insertion/deletion polymorphism. Anal. Biochem. 2010, 398, 126–128. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Chung, W.K. Quantitative analysis of copy number variants based on real-time LightCycler PCR. Curr. Protoc. Hum. Genet. 2014, 80. [Google Scholar] [CrossRef] [PubMed]
- Mazaika, E.; Homsy, J. Digital Droplet PCR: CNV Analysis and Other Applications. Curr. Protoc. Hum. Genet. 2014, 82, 7–24. [Google Scholar] [CrossRef]
- Armour, J.A.; Sismani, C.; Patsalis, P.C.; Cross, G. Measurement of locus copy number by hybridisation with amplifiable probes. Nucleic Acids Res. 2000, 28, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Patsalis, P.C.; Kousoulidou, L.; Sismani, C.; Männik, K.; Kurg, A. MAPH: From gels to microarrays. Eur. J. Med. Genet. 2005, 48, 241–249. [Google Scholar] [CrossRef]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.-J.; Chen, Z.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef]
- DNA Sequencing Costs: Data. Available online: https://www.genome.gov/about-genomics/fact-sheets/DNA-Sequencing-Costs-Data (accessed on 21 September 2020).
- Wang, H.; Nettleton, D.; Ying, K. Copy number variation detection using next generation sequencing read counts. BMC Bioinform. 2014, 15, 109. [Google Scholar] [CrossRef]
- Kosugi, S.; Momozawa, Y.; Liu, X.; Terao, C.; Kubo, M.; Kamatani, Y. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol. 2019, 20, 117. [Google Scholar] [CrossRef]
- Magi, A.; Bolognini, D.; Bartalucci, N.; Mingrino, A.; Semeraro, R.; Giovannini, L.; Bonifacio, S.; Parrini, D.; Pelo, E.; Mannelli, F.; et al. Nano-GLADIATOR: Real-time detection of copy number alterations from nanopore sequencing data. Bioinformatics 2019, 35, 4213–4221. [Google Scholar] [CrossRef]
- Bartha, Á.; Győrffy, B. Comprehensive Outline of Whole Exome Sequencing Data Analysis Tools Available in Clinical Oncology. Cancers 2019, 11, 1725. [Google Scholar] [CrossRef]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Mikheyev, A.S.; Tin, M.M.Y. A first look at the Oxford Nanopore MinION sequencer. Mol. Ecol. Resour. 2014, 14, 1097–1102. [Google Scholar] [CrossRef]
- Tham, C.Y.; Tirado-Magallanes, R.; Goh, Y.; Fullwood, M.J.; Koh, B.T.H.; Wang, W.; Ng, C.H.; Chng, W.J.; Thiery, A.; Tenen, D.G.; et al. NanoVar: Accurate characterization of patients’ genomic structural variants using low-depth nanopore sequencing. Genome Biol. 2020, 21, 56. [Google Scholar] [CrossRef] [PubMed]
- Lucas Lledó, J.I.; Cáceres, M. On the power and the systematic biases of the detection of chromosomal inversions by paired-end genome sequencing. PLoS ONE 2013, 8, e61292. [Google Scholar] [CrossRef] [PubMed]
- Buermans, H.P.J.; Den Dunnen, J.T. Next generation sequencing technology: Advances and applications. Biochim. Biophys. Acta 2014, 1842, 1932–1941. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, J.; Zhang, C.; Li, D.; Carvalho, C.M.B.; Ji, H.; Xiao, J.; Wu, Y.; Zhou, W.; Wang, H.; et al. Efficient CNV breakpoint analysis reveals unexpected structural complexity and correlation of dosage-sensitive genes with clinical severity in genomic disorders. Hum. Mol. Genet. 2017, 26, 1927–1941. [Google Scholar] [CrossRef]
- Kubiritova, Z.; Gyuraszova, M.; Nagyova, E.; Hyblova, M.; Harsanyova, M.; Budis, J.; Hekel, R.; Gazdarica, J.; Duris, F.; Kadasi, L.; et al. On the critical evaluation and confirmation of germline sequence variants identified using massively parallel sequencing. J. Biotechnol. 2019, 298, 64–75. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Kucharik, M.; Gnip, A.; Hyblova, M.; Budis, J.; Strieskova, L.; Harsanyova, M.; Pös, O.; Kubiritova, Z.; Radvanszky, J.; Minarik, G.; et al. Non-invasive prenatal testing (NIPT) by low coverage genomic sequencing: Detection limits of screened chromosomal microdeletions. PLoS ONE 2020, 15, e0238245. [Google Scholar] [CrossRef]
- Straver, R.; Sistermans, E.A.; Holstege, H.; Visser, A.; Oudejans, C.B.M.; Reinders, M.J.T. WISECONDOR: Detection of fetal aberrations from shallow sequencing maternal plasma based on a within-sample comparison scheme. Nucleic Acids Res. 2013, 42, e31. [Google Scholar] [CrossRef]
- Raman, L.; Dheedene, A.; De Smet, M.; Van Dorpe, J.; Menten, B. WisecondorX: Improved copy number detection for routine shallow whole-genome sequencing. Nucleic Acids Res. 2018, 47, 1605–1614. [Google Scholar] [CrossRef] [PubMed]
- Sathirapongsasuti, J.F.; Lee, H.; Horst, B.A.J.; Brunner, G.; Cochran, A.J.; Binder, S.; Quackenbush, J.; Nelson, S.F. Exome sequencing-based copy-number variation and loss of heterozygosity detection: ExomeCNV. Bioinformatics 2011, 27, 2648–2654. [Google Scholar] [CrossRef] [PubMed]
- Laver, T.W.; De Franco, E.; Johnson, M.B.; Patel, K.; Ellard, S.; Weedon, M.N.; Flanagan, S.E.; Wakeling, M.N. SavvyCNV: Genome-wide CNV calling from off-target reads. BioRxiv 2019, 617605. [Google Scholar] [CrossRef]
- Kuilman, T.; Velds, A.; Kemper, K.; Ranzani, M.; Bombardelli, L.; Hoogstraat, M.; Nevedomskaya, E.; Xu, G.; De Ruiter, J.; Lolkema, M.P.; et al. CopywriteR: DNA copy number detection from off-target sequence data. Genome Biol. 2015, 16, 49. [Google Scholar] [CrossRef]
- Fowler, A.; Mahamdallie, S.; Ruark, E.; Seal, S.; Ramsay, E.; Clarke, M.; Uddin, I.; Wylie, H.; Strydom, A.; Lunter, G.; et al. Accurate clinical detection of exon copy number variants in a targeted NGS panel using DECoN. Wellcome Open Res. 2016, 1, 20. [Google Scholar] [CrossRef]
- Talevich, E.; Hunter Shain, A.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef]
- Ivakhno, S.; Roller, E.; Colombo, C.; Tedder, P.; Cox, A.J. Canvas SPW: Calling de novo copy number variants in pedigrees. Bioinformatics 2018, 34, 516–518. [Google Scholar] [CrossRef]
- Zhao, H.; Huang, T.; Li, J.; Liu, G.; Yuan, X. MFCNV: A New Method to Detect Copy Number Variations from Next-Generation Sequencing Data. Front. Genet. 2020, 11, 434. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef]
- Amarasinghe, K.C.; Li, J.; Hunter, S.M.; Ryland, G.L.; Cowin, P.A.; Campbell, I.G.; Halgamuge, S.K. Inferring copy number and genotype in tumour exome data. BMC Genom. 2014, 15, 732. [Google Scholar] [CrossRef]
- Miller, C.A.; Hampton, O.; Coarfa, C.; Milosavljevic, A. ReadDepth: A parallel R package for detecting copy number alterations from short sequencing reads. PLoS ONE 2011, 6, e16327. [Google Scholar] [CrossRef]
- Chrisamiller Chrisamiller/Copycat. Available online: https://github.com/chrisamiller/copyCat (accessed on 6 January 2021).
- Yuan, X.; Bai, J.; Zhang, J.; Yang, L.; Duan, J.; Li, Y.; Gao, M. CONDEL: Detecting Copy Number Variation and Genotyping Deletion Zygosity from Single Tumor Samples Using Sequence Data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 17, 1141–1153. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Yu, J.; Xi, J.; Yang, L.; Shang, J.; Li, Z.; Duan, J. CNV_IFTV: An isolation forest and total variation-based detection of CNVs from short-read sequencing data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019. [Google Scholar] [CrossRef] [PubMed]
- Boeva, V.; Popova, T.; Bleakley, K.; Chiche, P.; Cappo, J.; Schleiermacher, G.; Janoueix-Lerosey, I.; Delattre, O.; Barillot, E. Control-FREEC: A tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 2012, 28, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Magi, A.; Tattini, L.; Cifola, I.; D’Aurizio, R.; Benelli, M.; Mangano, E.; Battaglia, C.; Bonora, E.; Kurg, A.; Seri, M.; et al. EXCAVATOR: Detecting copy number variants from whole-exome sequencing data. Genome Biol. 2013, 14, R120. [Google Scholar] [CrossRef]
- D’Aurizio, R.; Pippucci, T.; Tattini, L.; Giusti, B.; Pellegrini, M.; Magi, A. Enhanced copy number variants detection from whole-exome sequencing data using EXCAVATOR2. Nucleic Acids Res. 2016, 44, e154. [Google Scholar] [CrossRef]
- Magi, A.; Pippucci, T.; Sidore, C. XCAVATOR: Accurate detection and genotyping of copy number variants from second and third generation whole-genome sequencing experiments. BMC Genom. 2017, 18, 747. [Google Scholar] [CrossRef]
- Martin, J.; Tammimies, K.; Karlsson, R.; Lu, Y.; Larsson, H.; Lichtenstein, P.; Magnusson, P.K.E. Copy number variation and neuropsychiatric problems in females and males in the general population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2019, 180, 341–350. [Google Scholar] [CrossRef]
- Martin, C.L.; Wain, K.E.; Oetjens, M.T.; Tolwinski, K.; Palen, E.; Hare-Harris, A.; Habegger, L.; Maxwell, E.K.; Reid, J.G.; Walsh, L.K.; et al. Identification of Neuropsychiatric Copy Number Variants in a Health Care System Population. JAMA Psychiatry 2020. [Google Scholar] [CrossRef]
- Park, K.-B.; Nam, K.E.; Cho, A.-R.; Jang, W.; Kim, M.; Park, J.H. Effects of Copy Number Variations on Developmental Aspects of Children with Delayed Development. Ann. Rehabil. Med. 2019, 43, 215–223. [Google Scholar] [CrossRef]
- Glessner, J.T.; Li, J.; Desai, A.; Palmer, M.; Kim, D.; Lucas, A.M.; Chang, X.; Connolly, J.J.; Almoguera, B.; Harley, J.B.; et al. CNV Association of Diverse Clinical Phenotypes from eMERGE reveals novel disease biology underlying cardiovascular disease. Int. J. Cardiol. 2020, 298, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Prestes, P.R.; Maier, M.C.; Charchar, F.J. DNA copy number variations—Do these big mutations have a big effect on cardiovascular risk? Int. J. Cardiol. 2020, 298, 116–117. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhang, J.; Liao, D.; Yang, L.; Wang, Y.; Hou, S. Association between C4, C4A, and C4B copy number variations and susceptibility to autoimmune diseases: A meta-analysis. Sci. Rep. 2017, 7, 42628. [Google Scholar] [CrossRef] [PubMed]
- Pereira, K.M.C.; Perazzio, S.; Faria, A.G.A.; Moreira, E.S.; Santos, V.C.; Grecco, M.; Da Silva, N.P.; Andrade, L.E.C. Impact of C4, C4A and C4B gene copy number variation in the susceptibility, phenotype and progression of systemic lupus erythematosus. Adv. Rheumatol. 2019, 59, 36. [Google Scholar] [CrossRef]
- Szilágyi, M.; Pös, O.; Márton, É.; Buglyó, G.; Soltész, B.; Keserű, J.; Penyige, A.; Szemes, T.; Nagy, B. Circulating Cell-Free Nucleic Acids: Main Characteristics and Clinical Application. Int. J. Mol. Sci. 2020, 21, 6827. [Google Scholar] [CrossRef]
- Pös, O.; Biró, O.; Szemes, T.; Nagy, B. Circulating cell-free nucleic acids: Characteristics and applications. Eur. J. Hum. Genet. 2018, 26, 937–945. [Google Scholar] [CrossRef]
- Heitzer, E.; Ulz, P.; Geigl, J.B.; Speicher, M.R. Non-invasive detection of genome-wide somatic copy number alterations by liquid biopsies. Mol. Oncol. 2016, 10, 494–502. [Google Scholar] [CrossRef]
- Ni, X.; Zhuo, M.; Su, Z.; Duan, J.; Gao, Y.; Wang, Z.; Zong, C.; Bai, H.; Chapman, A.R.; Zhao, J.; et al. Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. Proc. Natl. Acad. Sci. USA 2013, 110, 21083–21088. [Google Scholar] [CrossRef]
- Pailler, E.; Auger, N.; Lindsay, C.R.; Vielh, P.; Islas-Morris-Hernandez, A.; Borget, I.; Ngo-Camus, M.; Planchard, D.; Soria, J.-C.; Besse, B.; et al. High level of chromosomal instability in circulating tumor cells of ROS1-rearranged non-small-cell lung cancer. Ann. Oncol. 2015, 26, 1408–1415. [Google Scholar] [CrossRef]
- Pös, Z.; Pös, O.; Styk, J.; Mocova, A.; Strieskova, L.; Budis, J.; Kadasi, L.; Radvanszky, J.; Szemes, T. Technical and Methodological Aspects of Cell-Free Nucleic Acids Analyzes. Int. J. Mol. Sci. 2020, 21, 8634. [Google Scholar] [CrossRef]
- Olsson, E.; Winter, C.; George, A.; Chen, Y.; Howlin, J.; Tang, M.-H.E.; Dahlgren, M.; Schulz, R.; Grabau, D.; Van Westen, D.; et al. Serial monitoring of circulating tumor DNA in patients with primary breast cancer for detection of occult metastatic disease. EMBO Mol. Med. 2015, 7, 1034–1047. [Google Scholar] [CrossRef]
- Peng, H.; Lu, L.; Zhou, Z.; Liu, J.; Zhang, D.; Nan, K.; Zhao, X.; Li, F.; Tian, L.; Dong, H.; et al. CNV Detection from Circulating Tumor DNA in Late Stage Non-Small Cell Lung Cancer Patients. Genes 2019, 10, 926. [Google Scholar] [CrossRef]
- Kubiritova, Z.; Radvanszky, J.; Gardlik, R. Cell-Free Nucleic Acids and their Emerging Role in the Pathogenesis and Clinical Management of Inflammatory Bowel Disease. Int. J. Mol. Sci. 2019, 20, 3662. [Google Scholar] [CrossRef]
- Pös, O.; Budiš, J.; Szemes, T. Recent trends in prenatal genetic screening and testing. F1000Res 2019, 8. [Google Scholar] [CrossRef]
- Pös, O.; Budis, J.; Kubiritova, Z.; Kucharik, M.; Duris, F.; Radvanszky, J.; Szemes, T. Identification of Structural Variation from NGS-Based Non-Invasive Prenatal Testing. Int. J. Mol. Sci. 2019, 20, 4403. [Google Scholar] [CrossRef]
- Giles, M.E.; Murphy, L.; Krstić, N.; Sullivan, C.; Hashmi, S.S.; Stevens, B. Prenatal cfDNA screening results indicative of maternal neoplasm: Survey of current practice and management needs. Prenat. Diagn. 2017, 37, 126–132. [Google Scholar] [CrossRef]
- Mohajeri, M.H.; Brummer, R.J.M.; Rastall, R.A.; Weersma, R.K.; Harmsen, H.J.M.; Faas, M.; Eggersdorfer, M. The role of the microbiome for human health: From basic science to clinical applications. Eur. J. Nutr. 2018, 57, 1–14. [Google Scholar] [CrossRef]
- Poole, A.C.; Goodrich, J.K.; Youngblut, N.D.; Luque, G.G.; Ruaud, A.; Sutter, J.L.; Waters, J.L.; Shi, Q.; El-Hadidi, M.; Johnson, L.M.; et al. Human Salivary Amylase Gene Copy Number Impacts Oral and Gut Microbiomes. Cell Host. Microbe. 2019, 25, 553–564. [Google Scholar] [CrossRef]
- Greenblum, S.; Carr, R.; Borenstein, E. Extensive strain-level copy-number variation across human gut microbiome species. Cell 2015, 160, 583–594. [Google Scholar] [CrossRef]
- Rice, A.M.; McLysaght, A. Dosage sensitivity is a major determinant of human copy number variant pathogenicity. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Gaziova, M.; Pos, O.; Krampl, W.; Kubiritova, Z.; Kucharik, M.; Radvanszky, J.; Budis, J.; Szemes, T. Automated prediction of the clinical impact of structural copy number variations. Bioinformatics 2020, 646. [Google Scholar] [CrossRef]
- Geoffroy, V.; Herenger, Y.; Kress, A.; Stoetzel, C.; Piton, A.; Dollfus, H.; Muller, J. AnnotSV: An integrated tool for structural variations annotation. Bioinformatics 2018, 34, 3572–3574. [Google Scholar] [CrossRef]
- Dharanipragada, P.; Vogeti, S.; Parekh, N. iCopyDAV: Integrated platform for copy number variations—Detection, annotation and visualization. PLoS ONE 2018, 13, e0195334. [Google Scholar] [CrossRef]
- AluScanCNV2: An R package for copy number variation calling and cancer risk prediction with next-generation sequencing data. Genes Dis. 2019, 6, 43–46. [CrossRef]
- Zhao, M.; Zhao, Z. CNVannotator: A Comprehensive Annotation Server for Copy Number Variation in the Human Genome. PLoS ONE 2013, 8, e80170. [Google Scholar] [CrossRef]
- Samarakoon, P.S.; Sorte, H.S.; Stray-Pedersen, A.; Rødningen, O.K.; Rognes, T.; Lyle, R. cnvScan: A CNV screening and annotation tool to improve the clinical utility of computational CNV prediction from exome sequencing data. BMC Genom. 2016, 17, 51. [Google Scholar] [CrossRef]
- Markham, J.F.; Yerneni, S.; Ryland, G.L.; Leong, H.S.; Fellowes, A.; Thompson, E.R.; De Silva, W.; Kumar, A.; Lupat, R.; Li, J.; et al. CNspector: A web-based tool for visualisation and clinical diagnosis of copy number variation from next generation sequencing. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Collins, R.L.; Stone, M.R.; Brand, H.; Glessner, J.T.; Talkowski, M.E. CNView: A visualization and annotation tool for copy number variation from whole-genome sequencing. bioRxiv 2016, 049536. [Google Scholar] [CrossRef]
- Ganel, L.; Abel, H.J.; Hall, I.M. SVScore: An impact prediction tool for structural variation. Bioinformatics 2017, 33, 1083–1085. [Google Scholar] [CrossRef]
- Erikson, G.A.; Deshpande, N.; Kesavan, B.G.; Torkamani, A. SG-ADVISER CNV: Copy-number variant annotation and interpretation. Genet. Med. 2015, 17, 714–718. [Google Scholar] [CrossRef]
- Spector, J.D.; Wiita, A.P. ClinTAD: A tool for copy number variant interpretation in the context of topologically associated domains. J. Hum. Genet. 2019, 64, 437–443. [Google Scholar] [CrossRef]
- Dalgleish, J.L.; Wang, Y.; Zhu, J.; Meltzer, P.S. CNVScope: Visually Exploring Copy Number Aberrations in Cancer Genomes. Cancer Inform. 2019, 18, 1176935119890290. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, Z.; Ban, R.; Zhang, H.; Iqbal, F.; Zhao, A.; Li, A.; Shi, Q. DeAnnCNV: A tool for online detection and annotation of copy number variations from whole-exome sequencing data. Nucleic Acids Res. 2015, 43, W289–W294. [Google Scholar] [CrossRef]
- Gurbich, T.A.; Ilinsky, V.V. ClassifyCNV: A tool for clinical annotation of copy-number variants. Sci. Rep. 2020, 10, 20375. [Google Scholar] [CrossRef]
- Acmg Board of Directors. Laboratory and clinical genomic data sharing is crucial to improving genetic health care: A position statement of the American College of Medical Genetics and Genomics. Genet. Med. 2017, 19, 721–722. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).