
Identification and Characterization of Known and Novel MicroRNAs in Five Tissues of Wax Gourd (Benincasa hispida) Based on High-Throughput Sequencing

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Small RNA Library Construction and Sequencing
2.3. Identification of miRNAs in Wax Gourd
2.4. Differential Expression Analysis of miRNAs
2.5. Target Gene Prediction, Functional Annotation and Pathway Analysis
2.6. Quantitative Real-Time PCR (qRT-PCR) Assay
3. Results
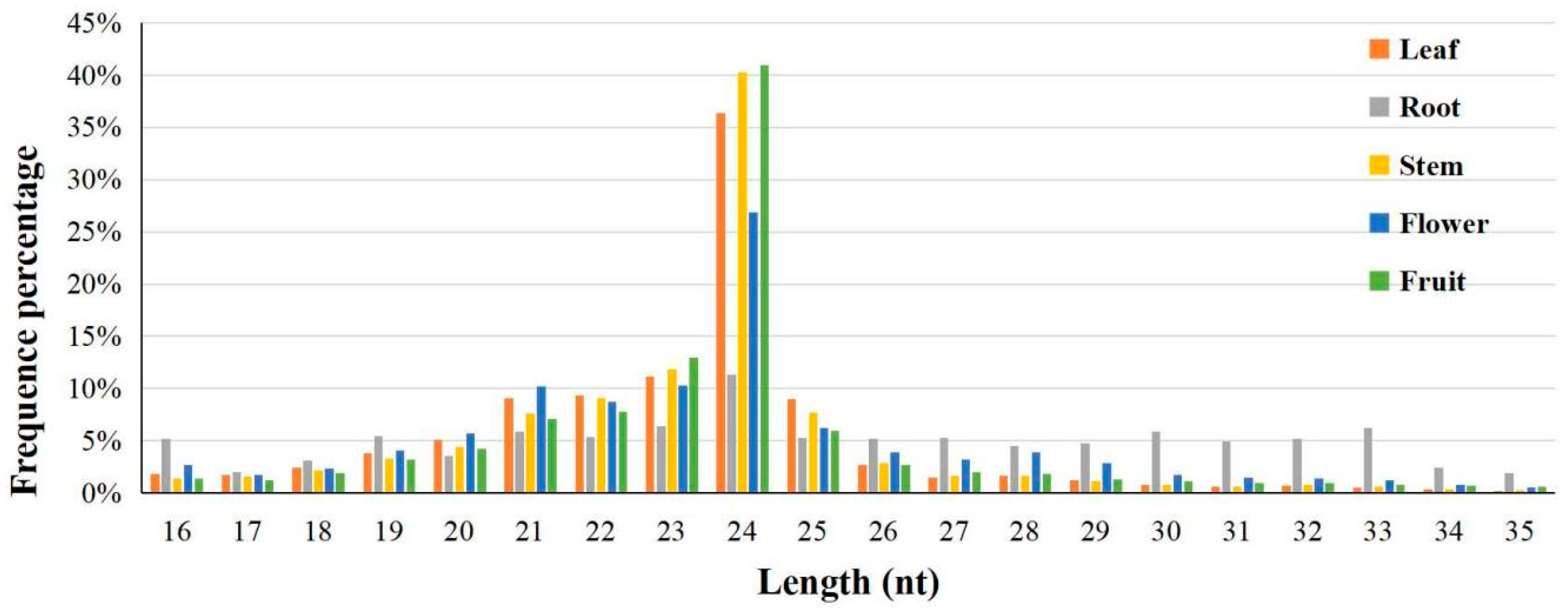
3.1. High-Throughput Sequencing and Analysis of Wax Gourd Small RNAs
3.2. Identification of Known miRNAs in Wax Gourd
3.3. Identification of Novel Potential Wax Gourd miRNAs
3.4. Validation of miRNAs Expression by qRT-PCR
3.5. Prediction of miRNA Targets in Wax Gourd
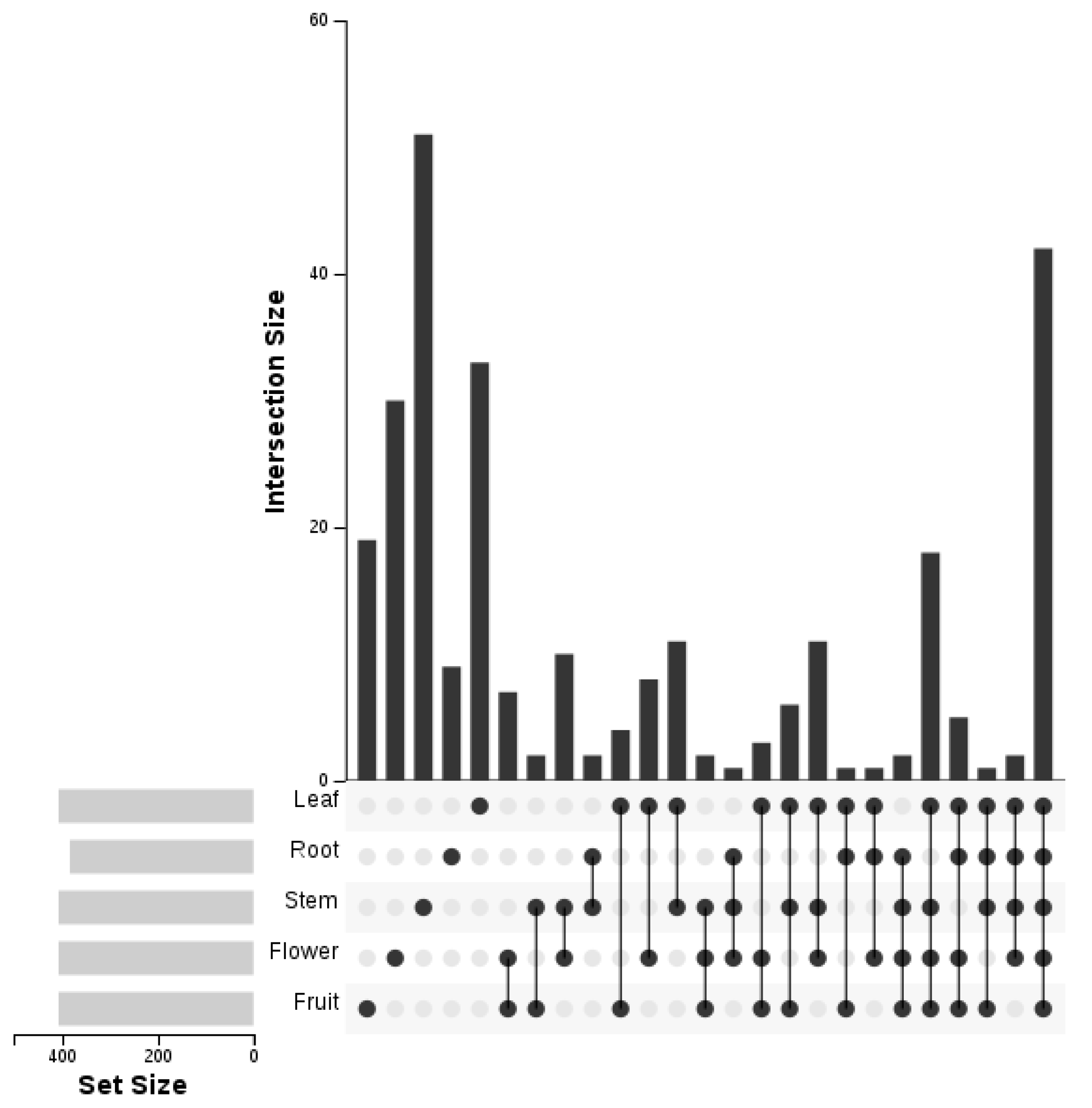
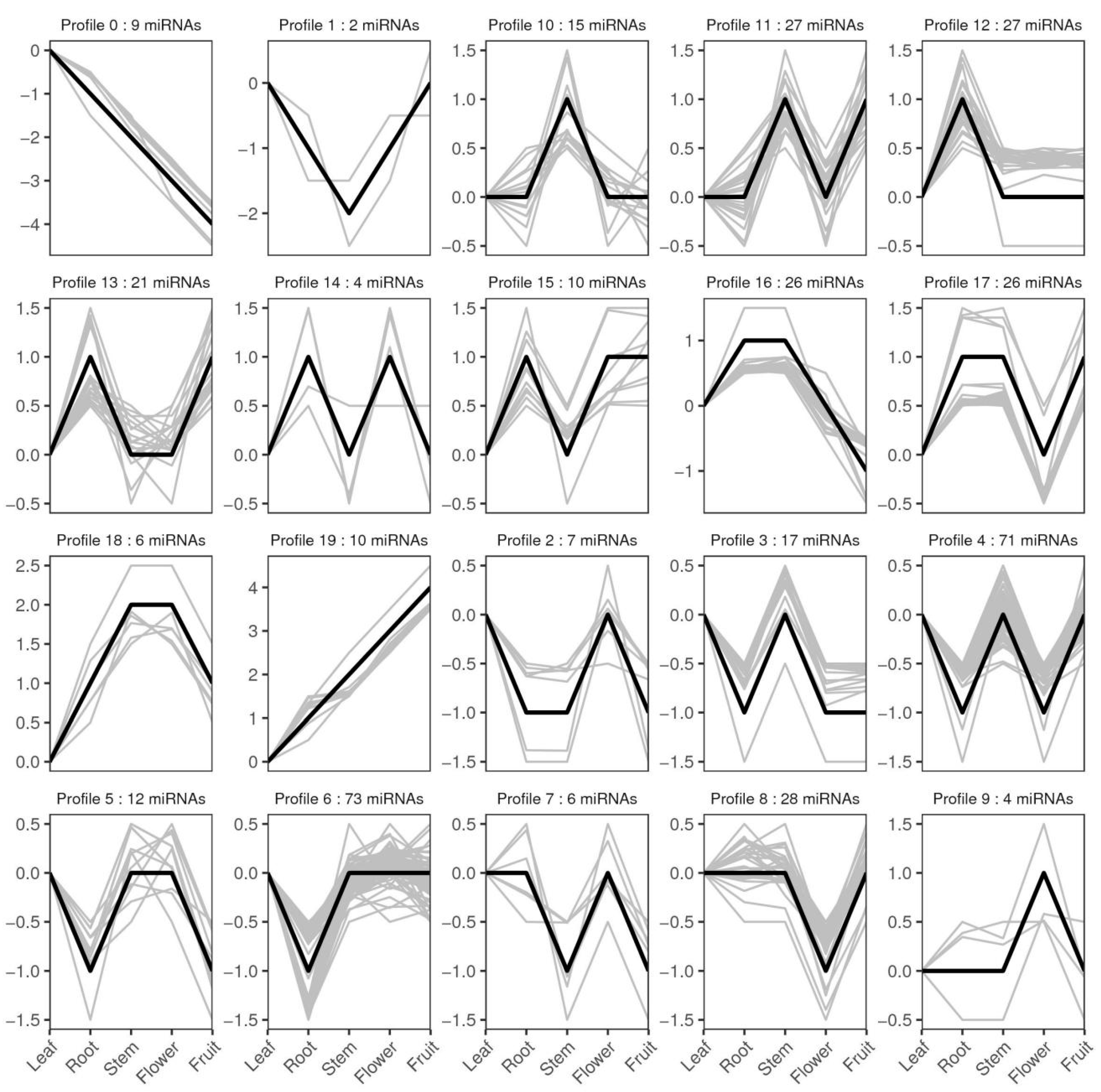
3.6. Differentially Expressed miRNAs between Five Libraries
3.7. Evolutionary Analysis of Wax Gourd miR164
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jones-Rhoades, M.W.; Bartel, D.P.; Bartel, B. MicroRNAs and their regulatory roles in plants. Annu. Rev. Plant Biol. 2006, 57, 19–53. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNA: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Millar, A.A.; Waterhouse, P.M. Plant and animal microRNAs: Similarities and differences. Funct. Integr. Genom. 2005, 5, 129–135. [Google Scholar] [CrossRef]
- Xie, Z.X.; Edwards, A.; Noah, F.; Adam, C.; Scott, A.G.; James, C.C. Expression of arabidopsis MiRNA genes. Plant Physiol. 2005, 138, 2145–2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, W.; Li, J.; Song, R.; Messing, J.; Chen, X. CARPEL FACTORY, a dicer homolog and HEN1, a novel protein, act in microRNA metabolism in Arabidopsis thaliana. Curr. Biol. 2002, 12, 1484–1495. [Google Scholar] [CrossRef] [Green Version]
- Kurihara, Y.; Watanabe, Y. Arabidopsis micro-RNA biogenesis through dicer-like 1 protein functions. Proc. Natl. Acad. Sci. USA 2004, 101, 12753–12758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Lu, Z.H. The fate of miRNA* strand through evolutionary analysis: Implication for degradation as merely carrier strand or protential regulatory molecule? PLoS ONE 2010, 5, e11387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, E.; Wuyts, J.; Rouz, P.; Van de Peer, Y. Detection of 91 potential conserved plant microRNAs in Arabidopsis thaliana and Oryza sativa identifies important target genes. Proc. Natl. Acad. Sci. USA 2004, 101, 11511–11516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.B.; Pan, X.P.; Wang, Q.L.; Cobb, G.P.; Anderson, T.A. Identification and characterization of new plant microRNAs using EST analysis. Cell Res. 2005, 15, 336–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Z.B.; Li, X.J.; Zhao, Y.Y.; Zhang, M.M.; Wan, Y.L.; Cao, D.C.; Lu, S.F.; Lin, J.X. Genome-wide analysis reveals dynamic changes in expression of micro RNAs during vascular cambium development in Chinese fir, Cunninghamialanceolata. J. Exp. Bot. 2015, 66, 3041–3054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Z.B.; Hai, B.Z.; Guo, J.L.; Li, Y.F.; Zhang, L. Characterization of wheat miRNAs and their target genes responsive to cadmium stress. Plant Physiol. Bioch. 2016, 101, 60–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saminathan, T.; Bodunrin, A.; Singh, N.V.; Devarajan, R.; Nimmakayala, P.; Jeff, M.; Aradhya, M.; Reddy, U.K. Genome-wide identification of microRNAs in pomegranate (Punicagranatum L.) by high-throughput sequencing. BMC Plant. Biol. 2016, 16, 122. [Google Scholar] [CrossRef] [Green Version]
- Shu, Y.J.; Liu, Y.; Li, W.; Song, L.L.; Zhang, J.; Guo, C.H. Genome-wide investigation of microRNAs and their targets in response to freezing stress in Medicago sativa L. based on high-throughput sequencing. G3 Genes Genomes Genet. 2016, 6, 755–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llave, C.; Xie, Z.; Kasschau, K.D.; Carrington, J.C. Cleavage of scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science 2002, 297, 2053–2056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barakat, A.; Sriram, A.; Park, J.; Zhebentyayeva, T.; Main, D.; Abbott, A. Genome wide identification of chilling responsive microRNAs in Prunus persica. BMC Genomics 2012, 13, 481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Q.Y.; Yang, C.Y.; Ma, Q.B.; Li, X.P.; Dong, W.W.; Nian, H. Identification of wild soybean miRNAs and their target genes responsive to aluminum stress. BMC Plant. Biol. 2012, 12, 182. [Google Scholar] [CrossRef] [Green Version]
- Baldrich, P.; Campo, S.; Wu, M.T.; Liu, T.T.; Hsing, Y.C.; San Segundo, B. MicroRNA-mediated regulation of gene expression in the response of rice plants to fungal elicitors. RNA Biol. 2015, 12, 847–863. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Yang, L.; Zeng, H.Q.; Zhou, Z.S.; Yang, Z.M.; Li, H.; Sun, D.; Xie, F.; Zhang, B. A cotton miRNA is involved in regulation of plant response to salt stress. Sci. Rep. 2016, 4, 6122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shriram, V.; Kumar, V.; Devarumath, R.; Khare, T.S.; Wani, S.H. MicroRNAs as potential targets for abiotic stress tolerance in plants. Front. Plant Sci. 2016, 7, 817. [Google Scholar] [CrossRef] [PubMed]
- Pagliarani, C.; Vitali, M.; Ferrero, M.; Vitulo, N.; Incarbone, M.; Lovisolo, C.; Valle, G.; Schubert, A. The accumulation of miRNAs differentially modulated by drought stress is affected by grafting in grapevine. Plant Physiol. 2017, 173, 2180–2195. [Google Scholar] [CrossRef] [Green Version]
- Huen, A.; Bally, J.; Smith, P. Identification and characterisation of microRNAs and their target genes in phosphate-starved Nicotiana benthamiana by small RNA deep sequencing and 5′RACE analysis. BMC Genom. 2018, 19, 940. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.Y.; Long, R.C.; Zhang, T.J.; Kang, J.M.; Wang, Z.; Wang, P.Q.; Sun, H.; Yu, J.; Yang, Q.C. Genome-wide identification of microRNAs in response to salt/alkali stress in Medicago truncatula through high-throughput sequencing. Int. J. Mol. Sci. 2018, 19, 4076. [Google Scholar] [CrossRef] [Green Version]
- Chitarra, W.; Pagliarani, C.; Abbà, S.; Boccacci, P.; Birello, G.; Rossi, M.; Palmano, S.; Marzachi, C.; Perrone, I.; Gambino, G. miRVIT: A novel miRNA database and its application to uncover Vitis responses to flavescence dorée infection. Front. Plant Sci. 2018, 9, 1034. [Google Scholar] [CrossRef]
- Juarez, M.T.; Kui, J.S.; Thomas, J.; Heller, B.A.; Timmermans, M.C. MicroRNA-mediated repression of rolled leaf1 specifies maize leaf polarity. Nature 2004, 428, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Nikovics, K.; Blein, T.; Peaucelle, A.; Ishida, T.; Morin, H.; Aida, M.; Laufs, P. The balance between the MIR164A and CUC2 genes controls leaf margin serration in Arabidopsis. Plant Cell 2006, 18, 2929–2945. [Google Scholar] [CrossRef] [Green Version]
- Berger, Y.; Harpaz-Saad, S.; Brand, A.; Melnik, H.; Sirding, N.; Alvarez, J.P.; Zinder, M.; Samach, A.; Eshed, Y.; Ori, N. The NAC-domain transcription factor GOBLET specifies leaflet boundaries in compound tomato leaves. Development 2009, 136, 823–832. [Google Scholar] [CrossRef] [Green Version]
- Bai, S.; Tian, Y.; Tan, C.; Bai, S.; Hao, J.; Hasi, A. Genome-wide identification of microRNAs involved in the regulation of fruit ripening and climacteric stages in melon (Cucumis melo). Hortic. Res. 2020, 7, 106. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Ortiz, C.; Peña-Garcia, Y.; Bhandari, M.; Abburi, V.L.; Natarajan, P.; Stommel, J.; Nimmakayala, P.; Reddy, U.K. Identification of miRNAs and their targets involved in flower and fruit development across domesticated and wild capsicum species. Int. J. Mol. Sci. 2021, 22, 4866. [Google Scholar] [CrossRef]
- Gupta, S.K.; Vishwakarma, A.; Kenea, H.D.; Galsurker, O.; Cohen, H.; Aharoni, A.; Arazi, T. CRISPR/Cas9 mutants of tomato MICRORNA164 genes uncover their functional specialization in development. Plant Physiol. 2021, kiab376. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Liu, W.R.; Xie, D.S.; Peng, Q.W.; He, X.M.; Lin, Y.E.; Liang, Z.J. High-density genetic map construction and gene mapping of pericarp color in wax gourd using specific-locus amplified fragment (SLAF) sequencing. BMC Genom. 2015, 16, 1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grover, J.K.; Adiga, G.; Vats, V.; Rathi, S.S. Extracts of Benincasa hispida prevent development of experimental ulcers. J. Ethnopharmacol. 2001, 78, 159–164. [Google Scholar] [CrossRef]
- Gu, M.; Fan, S.; Liu, G.; Guo, L.; Ding, X.; Lu, Y.; Zhang, Y.; Ji, G.; Huang, C. Extract of wax gourd peel prevents high-fat diet-induced hyperlipidemia in C57BL/6 mice via the inhibition of the PPARγ pathway. Evid. Based Complement. Alternat. Med. 2013, 2013, 342561. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.S.; Xu, Y.C.; Wang, J.P.; Liu, W.R.; Zhou, Q.; Luo, S.B.; Huang, W.; He, X.M.; Li, Q.; Peng, Q.W.; et al. The wax gourd genomes offer insights into the genetic diversity and ancestral cucurbit karyotype. Nat. Commun. 2019, 10, 5158. [Google Scholar] [CrossRef] [PubMed]
- Gardner, P.P.; Daub, J.; Tate, J.; Moore, B.L.; Osuch, I.H.; Griffiths-Jones, S.; Finn, R.D.; Nawrocki, E.P.; Kolbe, D.L.; Eddy, S.R.; et al. Rfam: Wikipedia, clans and the “decimal” release. Nucleic Acids Res. 2011, 39, D141–D145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Meyers, B.C.; Axtell, M.J.; Bartel, B.; Bartel, D.P.; Baulcombe, D.; Bowman, J.L.; Cao, X.; Carrington, J.C.; Chen, X.; Green, P.J.; et al. Criteria for annotation of plant microRNAs. Plant Cell 2008, 20, 3186–3190. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Zhao, P.X. PsRNA Target: A plant small RNA target analysis server. Nucleic Acids Res. 2011, 39, 155–159. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEEG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, 480–484. [Google Scholar] [CrossRef]
- Ambros, V.; Bartel, B.; Bartel, D.P.; Burge, C.B.; Carrington, J.C.; Chen, X.; Dreyfuss, G.; Eddy, S.R.; Griffiths-Jones, S.; Marshall, M.; et al. A uniform system for microRNA annotation. RNA 2003, 9, 277–279. [Google Scholar] [CrossRef] [Green Version]
- Reinhart, B.J.; Weinstein, E.G.; Rhoades, M.W.; Bartel, B.; Bartel, D.P. MicroRNAs in plants. Gene Dev. 2002, 16, 1616–1626. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Hannom, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 631. [Google Scholar] [CrossRef] [Green Version]
- Chen, X. Small RNAs and their roles in plant development. Annu. Rev. Cell Dev. B 2009, 25, 21–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J.H.; Seo, P.J.; Park, C.M. MicroRNA biogenesis and function in higher plants. Biotechnol. Rep. 2009, 3, 111–126. [Google Scholar] [CrossRef]
- Li, B.S.; Qin, Y.R.; Duan, H.; Yin, W.L.; Xia, X.L. Genome-wide characterization of new and drought stress responsive microRNAs in Populus euphratica. J. Exp. Bot. 2011, 62, 3765–3779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, W.H.; Li, Z.Y.; Xia, X.J.; Li, Y.D.; Yu, Q.J. A combined approach of high-throughput sequencing and degradome analysis reveals tissue specific expression of microRNAs and their targets in cucumber. PLoS ONE 2012, 7, e33040. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.D.; Liu, L.B.; Liu, L.F.; Li, H.Y.; Zhang, Y.H.; Yao, Y.Y.; Ni, Z.F.; Gao, J.W. High-throughput sequencing discovery of conserved and novel microRNAs in Chinese cabbage (Brassica rapa L. ssp. pekinensis). Mol. Genet. Genomic. 2012, 287, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wang, Y.; Xu, Y.Y.; Wang, L.J.; Zhai, L.L.; Zhu, X.W.; Gong, Y.Q.; Ye, S.; Liu, L.W. Identification and characterization of novel and conserved microRNAs in radish (Raphanus sativus L.) using high-throughput sequencing. Plant Sci. 2013, 201–202, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.J.; Zhang, X.N.; Luo, Z.R.; Zhang, Q.L.; Liu, J.H. Identification and characterization of microRNAs from Chinese pollination constant non-astringent persimmon using high-throughput sequencing. BMC Plant Biol. 2015, 15, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, H.; Zhang, L.B.; Wu, G.; Fu, C.H.; Long, Y.; Xiang, J.; Gan, J.P.; Zhou, Y.H.; Yu, L.J.; Li, M.T. Genome-wide identification and characterization of microRNAs and target genes in Lonicera japonica. PLoS ONE 2016, 11, e0164140. [Google Scholar] [CrossRef]
- Bouchareb, A.; Le Cam, A.; Montfort, J.; Gay, S.; Nguyen, T.; Bobe, J.; Thermes, V. Genome-wide identification of novel ovarian-predominant miRNAs: New insights from the medaka (Oryzias latipes). Sci. Rep. 2017, 7, 40241. [Google Scholar] [CrossRef] [PubMed]
- Martínez, G.; Forment, J.; Llave, C.; Pallás, V.; Gómez, G. High-throughput sequencing, characterization and detection of new and conserved cucumber mirnas. PLoS ONE 2011, 6, e19523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Leng, X.P.; Zhang, Y.Y.; Kayesh, E.; Zhang, Y.P.; Sun, X.; Fang, J.G. Transcriptome-wide analysis of dynamic variations in regulation modes of grapevine microRNAs on their target genes during grapevine development. Plant Mol. Biol. 2014, 84, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; Lu, X.; Lian, C.L.; An, Y.; Xia, X.L.; Yin, W.L. Genome-wide analysis of microRNA responses to the phytohormone abscisic acid in Populuseuphratica. Front. Plant Sci. 2016, 7, 1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez, F. Arabidopsis endogenous small RNAs: Highways and byways. Trends Plant Sci. 2006, 1, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Herr, A.J. Pathways through the small RNA world of plants. FEBS Lett. 2005, 579, 5879–5888. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.T.; Hu, S.H.; Yan, C.X.; Li, C.J.; Zhao, X.B.; Wan, S.B.; Shan, S.H. Mining, identification and function analysis of microRNAs and target genes in peanut (Arachishypogaea L.). Plant Physiol. Bioch. 2017, 111, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, L.; Chen, D.; Wu, X.; Huang, D.; Chen, L.; Li, L.; Deng, X.; Xu, Q. Genome-wide comparison of microRNAs and their targeted transcripts among leaf, flower and fruit of sweet orange. BMC Genom. 2014, 15, 695. [Google Scholar] [CrossRef] [Green Version]
- Rosas-Cárdenas, F.d.F.; Caballero-Pérez, J.; Gutiérrez-Ramos, X.; Marsch-Martínez, N.; Cruz-Hernández, A.; de Folter, S. miRNA expression during prickly pear cactus fruit development. Planta 2015, 241, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Larue, C.T.; Wen, J.; Walker, J.C. A microRNA-transcription factor module regulates lateral organ size and patterning in Arabidopsis. Plant J. 2009, 58, 450–463. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue | Raw Reads | Clean Reads | 3′ Adapter | Insert | 5′ Adapter | Poly A | Clean Tags |
|---|---|---|---|---|---|---|---|
| Leaf | 19,026,019 | 18,675,941 | 264,840 | 71,345 | 14,955 | 1704 | 18,284,736 |
| Root | 20,596,286 | 20,161,705 | 244,410 | 37,464 | 22,930 | 699 | 19,813,111 |
| Stem | 20,335,677 | 19,947,266 | 270,636 | 68,005 | 14,962 | 1773 | 19,551,447 |
| Flower | 23,542,034 | 23,155,945 | 219,221 | 71,716 | 17,647 | 1279 | 22,821,341 |
| Fruit | 16,351,738 | 16,085,205 | 156,295 | 51,076 | 10,107 | 1253 | 15,843,596 |
| Leaf | Root | Stem | Flower | Fruit | |
|---|---|---|---|---|---|
| Known miRNAs | 255 | 146 | 264 | 219 | 213 |
| Novel miRNAs | 407 | 381 | 408 | 408 | 405 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, J.; Wang, M.; Liu, W.; Xie, D.; He, X.; Peng, Q.; Jiang, B. Identification and Characterization of Known and Novel MicroRNAs in Five Tissues of Wax Gourd (Benincasa hispida) Based on High-Throughput Sequencing. Appl. Sci. 2021, 11, 10068. https://doi.org/10.3390/app112110068
Yan J, Wang M, Liu W, Xie D, He X, Peng Q, Jiang B. Identification and Characterization of Known and Novel MicroRNAs in Five Tissues of Wax Gourd (Benincasa hispida) Based on High-Throughput Sequencing. Applied Sciences. 2021; 11(21):10068. https://doi.org/10.3390/app112110068
Chicago/Turabian StyleYan, Jinqiang, Min Wang, Wenrui Liu, Dasen Xie, Xiaoming He, Qingwu Peng, and Biao Jiang. 2021. "Identification and Characterization of Known and Novel MicroRNAs in Five Tissues of Wax Gourd (Benincasa hispida) Based on High-Throughput Sequencing" Applied Sciences 11, no. 21: 10068. https://doi.org/10.3390/app112110068