1. Introduction
Diatom algae are a major primary producer in the biosphere, fixating approximately 20% of all organic carbon on the planet. They have also been used in a number of biotechnological applications, including biofuel production [
1], biosynthesis of various pharmaceuticals and food supplements [
2], drug delivery systems [
3,
4], biodegradation of toxic compounds [
5], fish feed [
6], and other applications recently reviewed in, e.g., [
7]. There is also a tantalizing promise made by diatom biosilification capability: these algae feature siliceous cell walls with finely patterned nanostructures. Should the mechanism behind the production of these cell walls be understood and reproduced, it would have a large impact on nano-scale engineering. Finally, genetic engineering has been successfully used on diatoms (see e.g., [
8] for a review of methods and applications), further expanding the range of their possible roles in biotechnology.
All these applications require an understanding of diatom metabolism and genetics that can be provided by genome sequencing studies. A number of such studies have been performed, some using model species [
9,
10] and others focusing on promising candidates for biofuel production [
1,
11]. One of these studies, co-authored by authors of this work, was done on
Fragilaria radians (Kützing) D.M. Williams & Round (=
Synedra acus subsp.
radians (Kützing) Skabichevskij) isolated from Lake Baikal, a major player in the lake ecosystem, and potentially a model freshwater diatom [
12].
Diatom algae in natural habitats are colonized by numerous bacteria that interact with their host in complex ways, ranging from simple consumption of diatom-produced polysaccharides to algicidal activity [
13,
14,
15]. This close relationship with bacteria has always been a feature of diatoms, as evidenced by a high amount of bacterium-derived genes in their genomes, most likely acquired via horizontal genetic transfer (HGT) throughout evolutionary history [
16]. On the other hand, genomic sequencing and other application may require the production of axenic biomass, which is possible in culture.
F. radians strains used in the genomic project were axenized using a previously developed protocol [
17]. Axenization strategy involved filtration through a polycarbonate membrane with a pore size of 5 μm (to remove free bacteria), detergent treatment (20 μg/mL Triton X-100), cell treatment with the selected antibiotic (5 μg/mL ciprofloxacin, 18 h incubation), repeated filtration, and monoclonal culturing of diatom cells. Axenic diatom culture was grown in DM medium [
18] in 100 mL Erlenmeyer flasks up to a density of 10
4 cells/mL and then moved to 20 L bottles for further biomass growth. Since the cultures could have been recolonized by bacteria at some point between axenization and sequencing, their purity was further controlled by light microscopy (using DAPI staining) right before DNA isolation, and by measuring the abundance of non-organellar 16S rRNA sequences in read libraries. Both of these methods have detected no contamination, and a number of libraries was produced using 454, Illumina HiSeq and PacBio sequencers. Later, these libraries were used to assemble, annotate, and eventually publish
F. radians genome [
12] and transcriptome [
19].
Two strains were used for whole-genome and transcriptome sequencing. Initially, strain G9 isolated from South Baikal was sequenced from 2010 to 2012 to produce a series of shotgun libraries on Roche 454 sequencer, and also used in 2013 to produce a single Illumina MiSeq 2 × 250 paired-end library. These data were used in the assembly that was published in [
12]. By that time, strain G9 was lab-cultured for close to 10 years, leading to decrease in cell size and viability. It was replaced with a new strain, dubbed Ax BK280, which was also isolated from South Baikal and axenized according to the same protocol. This new strain was used to produce one library on PacBio RS2 in 2015, one library on Illumina HiSeq in 2016, and a series of transcriptomic libraries in 2019. Genomic libraries from strain Ax BK280 were not included into any published assemblies.
However, the precautions taken during genome sequencing were not sufficient. Large-scale bacterial contamination was later detected by a study focusing on HGT process in diatoms [
16]. Whereas normally HGT-derived genes would be distributed broadly both along the genome and along the taxonomy of donor bacteria,
F. radians assembly was found to contain whole scaffolds derived from
Sphingomonas sp. Further, the GC-content of these scaffolds is much closer to
Sphingomonas genomes than it is to the rest of
F. radians genome. The authors of that study considered them artifactual and suggested that the culture was, in fact, contaminated by
Sphingomonas sp.
Thus, our goals in this work were threefold: first, to establish whether there are any non-contaminated libraries in our dataset; second, to find out when has the contamination happened and whether the existing cultures were axenic; and the last but not the least, to document the flaws in our methods of detecting contamination.
2. Materials and Methods
Existing genomic and transcriptomic reads were mapped to the published
F. radians genomic assembly [
12] using bowtie2 2.3.0 [
20] for 454 and Illumina reads, and BLASR [
21] for PacBio reads. All transcriptomic libraries produced from strain Ax BK280 were pooled together for this work. Bowtie mapping was performed on default settings, except for insert sizes in mate-pair and paired-end libraries: 2–5 Kbp for library 14092013, 200–800 bp for library 27022013, and 250–700 bp for library A280-Illumina. BLASR mapping was performed on default settings.
16S rRNA reads were extracted from all libraries by aligning a whole library to SILVA v138 reference rRNA alignment in mothur 1.41 [
22]. All reads that successfully aligned were classified using mothur’s naive Bayesian classifier. To visualize the distribution of these reads along the 16S rRNA gene, a proportion of non-gap characters in the alignment of reads to the reference alignment, normalized between libraries, was plotted along the length of this reference. All statistical analyses and visualizations were performed in Python 3.6 using Biopython and Pyplot.
Diatom cultures of strain AxBK280 were grown in DM medium (V 600 mL) in a incubator at 8–10 °C and 16 μmol/m
2·s light intensity with 12:12 day:night cycle. For DNA isolation, the cells were grown up to a density of 10
4 cells/mL. For the 16S rRNA sequencing, DNA was isolated from diatom cultures according to the protocol available at [
http://dx.doi.org/10.17504/protocols.io.qh6dt9e, last accessed 27 October 2021]. The V4 variable region of the 16S rRNA gene was amplified using universal bacterial primers (F515 5′-GTGCCAGCMGCCGCGGTAA-3′ and R806 5′-GGACTACVSGGGTATCTAAT-3′) and sequenced on Illumina MiSeq. PCR amplification and sequencing were performed at the Core Centrum “Genomic Technologies, Proteomics and Cell Biology” in ARRIAM (All-Russia Research Institute for Agricultural Microbiology, St. Petersburg, Russia). Samples from 2020 and 2021 were extracted from fresh culture sedimented by centrifugation at 2500×
g, +4 °C. DNA from 2017 was extracted in 2020 from frozen culture samples, which were sedimented similarly and stored at −70 °C.
Amplicon libraries were analyzed in mothur 1.41 with SILVA v138.1 reference aligment downloaded from mothur’s website. The completeness of
Sphingomonas genome in
F. radians genomic assembly was estimated using BUSCO v5 [
23] on gVolante web server [
24].
For the phylogenetic analysis of
Sphingomonas 16S rRNA we have extracted 16S gene from the genomic assembly and constructed 90% majority rule consensus sequences for all amplicon OTUs classified as
Sphingomonas. We have also included the sequence of
F. radians-associated
Sphingomonas sp. described in [
25]. All representatives of genera
Sphingomonas and
Novosphingobium were extracted from SILVA v138.1; 200 were selected for the analysis using DJ sampling method [
26] with Kimura distance. Genus
Novosphingobium was included in this analysis because its member was described in [
25] as a closest BLAST hit for the sequence of
Sphingomonas sp. All
F. radians-associated sequences were aligned to a subset of SILVA alignment using Clustal Omega [
27]. Alignment was further corrected manually. ML phylogenetic tree was built using IQtree [
28] on both complete alignment and its subset trimmed by amplicon boundaries.
3. Results
The list of genes and scaffolds thought to contain Sphingomonas contamination was generously provided by Emmelien Vancaester. In total, this list includes 3644 genes on 697 scaffolds with total length of approx. 4 Mbp. Their results confirm high sequence identity and similar GC-content between these scaffolds and genomes of various Sphingomonadales, but that does not automatically imply contamination. Although a huge wave of HGTs from Sphingomonas to F. radians is unlikely, it is not outright impossible. To validate that these scaffolds are not, in fact, valid F. radians genes, we have mapped available transcriptomic reads on the whole genome assembly. The reasoning behind this is that the cDNA used in transcriptomic project was amplified using oligo(dT) primers that bind polyadenylated eukaryotic mRNA. Sphingomonas mRNA, lacking polyadenylation, would thus be underrepresented or even completely absent from transcriptomic dataset. Therefore, if the scaffolds in question are poorly covered by the transcriptomic data, it would serve as further evidence of their bacterial origin.
As
Figure 1 shows, transcriptomic coverage of the supposedly
Sphingomonas-derived scaffolds is extremely low. Only a few genes show any coverage at all, while the majority are completely absent from transcriptomic data.
Absence of these genes from transcriptomic data shows that they are not a part of F. radians active gene repertoire. However, this does not necessarily imply that they are not a part of its genome. There are at least two alternative interpretations: they may be valid F. radians genes not expressed under conditions of our transcriptomic experiment, or they may be pseudogenized. It is also possible that these genes are indeed bacterial, but the contamination was not present in samples used for RNA-seq.
According to [
16], the scaffolds that contain these genes show remarkable sequence similarity to Sphingomonadales (the authors used maximum likelihood phylogeny to detect origins, and later filtered contigs using BLAST hits to Sphingomonadales with >75% identity and >25% contig coverage) and higher GC-content (63.3% vs. genome average of 42.1%,
p < 2 × 10
−16, see
Figure 2b in [
16] for a more detailed view). Further, no other diatoms were found to contain that many HGT-derived genes acquired from a single source and not shared with at least one other diatom. BUSCO analysis of these scaffolds shows that they include 89.59% of the Sphingomonadales ortholog set (84.58% complete, 5.01% partial). In other words, this nearly complete ortholog set seems to have been sampled from a bacterial genome, which is to be expected if we have sequenced a mixture of
F. radians and
Sphingomonas sp. cells.
Pseudogenization or low expression levels fail to explain these observations, so we can safely assume that genes identified as belonging to Sphingomonas are, in fact, a product of bacterial contamination.
To establish whether this contamination persists in all sequencing data, whole-genome sequencing libraries were mapped to the published assembly. The number and proportion of reads mapped to
Sphingomonas-derived scaffolds are shown in
Table 1. For reference, these scaffolds comprise 4 Mbp of the assembly 98.4 Mbp in size, or ~4.07% of the total assembly length.
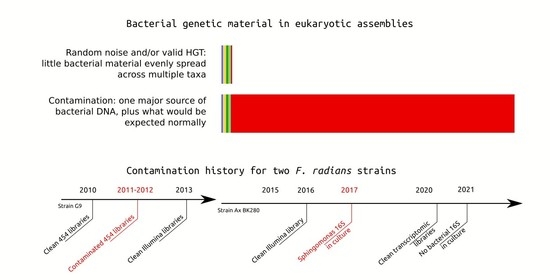
These numbers show that reads mapping to the scaffolds in question are overrepresented in 454 libraries from 2011–2012, suggesting that corresponding DNA was present in these samples but not the rest. This is a further argument towards the bacterial origin of these scaffolds and a hint as to when the contamination happened. It is important to note that we considered whole scaffolds to be bacterial, although it is possible that some of them are chimeric, i.e., include fragments from genomes of both F. radians and Sphingomonas sp.
This suspicion is further confirmed by taxonomic analysis of 16S rRNA reads extracted from these libraries. In case of contaminated libraries, it points to Sphingomonas, or at least unclassified Sphingomonadales, as a major source of bacterial rRNA. In other libraries, with a single exception, there are no more than a few reads confidently classified as something other than mitochondria or chloroplasts. The exception is library GKAKADZ02; although it appears to be “clean” based on a number of reads mapping to Sphingomonas scaffolds, this library contains a large number of 16S rRNA reads derived from diverse bacteria.
The origin of these reads can be clarified by plotting the coverage of SILVA reference alignment by non-organellar reads from this library (
Figure 2; distribution for library GKAKADZ02 is shown in red). For all other libraries, this plot shows a roughly U-shaped curve with high coverage of alignment ends and a relatively low coverage in the middle. This shape can be explained by the fact that this alignment contains multiple gaps in the 6000–35,000 region including V2–V6 16S rRNA hypervariable regions, so each read is stretched over multiple kilobases of alignment length, producing lower per-base coverage. For library GKAKADZ02, though, the plot suggests that a huge amount of reads covering a very specific region (roughly corresponding to V2-V5 variable regions of rRNA, a segment commonly used in 16S rRNA-based diversity studies) was added to a normal shotgun library. Together with an elevated bacterial diversity this result suggests contamination by an SSU amplicon during storage or sequencing run preparation.
Analysis of shotgun libraries has shown that
Sphingomonas genomic DNA was only present in the libraries produced from strain G9 during 2011–2012. Later libraries produced from this strain, as well as all libraries produced from strain Ax BK280, appear to be clean. However, we have decided to further validate the purity of Ax BK280 using 16S rRNA amplicon sequencing. Insufficient sensitivity of light microscopy with DAPI staining was already shown by the fact that bacterial contamination went undetected in 2011–2012, while DNA-based methods are thought to produce false negatives (i.e., detect no contamination when it is present) only in rare cases [
29,
30].
Three DNA samples were isolated from strain Ax BK280 using frozen cells from March 2017 and fresh culture produced in November 2020 and February 2021. Complete list of taxonomic assignments for generated OTUs is available in
Supplementary Table S1. However, the essential result is that the samples from 2020 and 2021 do not contain any abundant bacteria at all. The reads that were identified as non-organelle bacteria are scattered among numerous singletons and small phylotypes with single-digit read counts. In our opinion, these represent noise (for comparison, rates of 0.02–0.06% of library for the bacteria known to be absent from sample were previously documented for MiSeq 16S rRNA amplicon sequencing [
31]), and the absence of any single major source of bacterial reads implies that the diatom culture is free from bacterial contamination. In contrast, the sample from 2017 contains
Sphingomonas as a single dominant phylotype (besides plastids), confirming that this strain was also contaminated by the same bacterium as strain G9, or at least its relative.
Thus, a series of 2011–2012 454 libraries of strain G9, as well as strain Ax BK280 in 2017, contain some
Sphingomonas DNA. The bacterium itself was never observed, despite using DAPI staining to monitor culture axenicity. Microphotographs of cells from corresponding cultures are shown in
Figure 3.
To find out whether the same bacterial strain was present in 2011–2012 and 2017, we have built ML phylogenetic trees including 4 OTU consensus sequences, 16S rRNA fragment extracted from genomic assembly, and
Sphingomonas sp. strain baik7s previously co-isolated with F. radians [
25]. Trees were built both using the complete alignment (
Supplementary Figure S1A), and only using positions corresponding to V3-V4 16S rRNA fragment boundaries sequenced from 2017 samples (
Figure 4,
Supplementary Figure S1B).
Both trees show that two contaminants represent different
Sphingomonas species. In the tree built on the positions limited to the amplicon boundaries (shown in simplified form in
Figure 4), three
Sphingomonas groups are scattered over the tree with multiple reference clades between them (all OTU consensi are sister to each other). In contrast, the tree built on complete alignment recovers contaminant from 2011–2012 as sister to strain baik7s, but these two leaves are shown as basal relative to the rest of the tree, and the former has a very long branch. Thus, we interpret this result as a long-branch attraction artifact probably caused by low-quality alignment, and conclude that three separate
Sphingomonas strains were colonizing
F. radians cultures at different times. Since most sequences in SILVA (as well as closest BLAST hits against NCBI nr) are environmental and lack taxonomic identification below genus, we do not make any claims regarding the precise taxonomy of both contaminants.
4. Discussion
The collected data show that two supposedly axenic Fragilaria radians strains had a very similar history. They were originally successfully axenized and kept in this state for years, only to be eventually contaminated by Sphingomonas sp. The strains were routinely checked for bacterial contamination using DAPI staining, and some contaminated samples were discarded, but Sphingomonas sp. was not detected (presumably due to extremely low counts). At some point the bacteria died out in both strains, although no additional axenization was undertaken. If it was low counts that helped the bacterium remain undetected, they could also have prevented it from being transmitted along with algal cells during passage.
It is also possible that Sphingomonas sp. never contaminated the culture itself, instead inhabiting some element of lab equipment. Despite the fact that the vials and the culture medium were sterilized for the cultivation of diatoms, we admit the possibility of bacterial contamination due to poor-quality sterilization of the equipment or contaminated reagents used in DNA extraction and library preparation.
Since no observation of either Sphingomonas sp. strain was ever made directly, any detailed discussion of how the algal culture was contaminated or decontaminated would be highly speculative. Yet, we suspect that large-volume growth pipeline may have been a main culprit. Up to 15 L of water for medium preparation were sterilized simultaneously in a high-pressure steam sterilizer at 121 °C, potentially creating insufficiently lethal conditions for some Sphingomonas strains. We also cannot exclude contamination during medium preparation, diatom inoculation or long-term culture growth (normally approx. 30 days). All these stages were carried in a room that was clean, but not sterile.
The high-volume growth may also have caused us to miss bacterial contamination in microscopy-based controls. Diatom cells were sedimented from the entire culturing volume on 5 μm filters, and approx. 50 μL of diatom biomass were sampled for the microscopy. If bacterial cells form aggregates in the culture, they may be unevenly distributed in this concentrated diatom biomass and thus not present in a small volume used for microscopy.
For these two reasons, we have decided to abandon high-volume axenic diatom culturing, instead growing cells in multiple 1000 μL flasks. Medium preparation and inoculation are carried out in sterile laminar boxes, and microscopy controls are made before and after biomass sedimentation.
The details of these strains’ contamination are not very interesting by themselves (except for revising F. radians genome assembly, which is beyond the scope of this paper), but this case demonstrates multiple ways in which contamination goes undetected. Although care was taken to keep the strains axenic, bacterial contamination was noticed neither in culture, nor in read libraries, nor in published genomic data. Thus, it would be useful to describe how these failures occurred and how they could’ve been avoided.
As mentioned above, light microscopy detected no bacterial cells during the entire lifetime of both strains, although each of them has probably contained bacteria for years. Since we do not have any hard evidence of why this happened, we cannot provide reliable guidelines for preventing such occurrences in the future. We can only suggest that microscopy should not be relied upon without corroborating evidence.
As noted before [
32], 16S amplicon sequencing can be a useful addition, as this method is relatively cheap and does not make any assumptions about the taxonomy of possible contaminants (as would PCR or RT-PCR used for this purpose in older works [
29]). It can even have some limited utility in detecting eukaryotic contaminants by recovering sequences of their plastids or mitochondria. On the other hand, this method requires a molecular biology laboratory and could be prone to false positives due to the contamination of DNA samples after isolation. It also requires careful interpretation: even in a clean sample, some reads would inevitably be classified as non-organellar bacteria due to a combination of in vitro amplification/sequencing artifacts and in silico classification errors. However, real contamination is likely to be caused by one bacterial strain, which will be represented by one or a few numerous phylotypes (or OTUs). Random noise, on the other hand, is likely to be distributed evenly along the bacterial taxonomy. There is also an issue of unclassified reads that could be coming from the contaminant bacterium, host organelles, amplification artifacts, or any other source.
Analysis of F. radians shotgun read libraries for bacterial sequences has produced both false negatives (missing the contamination when it is present) and false positives (finding contamination in clean libraries). False positive due to contamination with a 16S amplicon should be a rare occasion; a high amount of bacterial 16S rRNA reads usually does imply contamination. Technically, a positive is not even false: the library has been contaminated, after all! Still, we believe that this case deserves a mention. Although the library shows high content of bacterial 16S rRNA, it is safe to use in genomic assembly. These reads are unlikely to map to anything other than SSU genes, and they are trivial to filter out. False negatives are harder to detect, as it is difficult to establish a baseline number of reads that should map to bacterial 16S rRNAs without a clean reference library. However, both types of errors could be avoided with careful examination of taxonomic distribution of recovered reads: overall approach should be similar to that described above for amplicon libraries.
The same advice applies to other bacterial sequences in eukaryotic genomic libraries. Most eukaryotes have at least a few HGT-derived genes, so the fact that some reads map to bacterial genomes does not constitute evidence of contamination. This is especially true for diatom algae, who are known to have high rates of horizontal gene acquisition [
10] and generally mosaic genome composition caused by multiple endosymbioses in their evolutionary history. Another complication is introduced by a relative scarcity and non-model status of diatom genomes. At least in the absence of high-quality reference genome, any seemingly bacterial read could be interpreted either as an HGT-derived gene or as a part of contaminant’s genome. However, real HGTs are relatively rare and involve various bacterial donors, while contamination introduces a single bacterial genome in its entirety into the library.
In case of protein-coding genes one could also rely on low-level statistics such as GC content or frequencies of short k-mers, as was done in [
16]. These parameters are relatively species-specific and differ significantly between eukaryotes and bacteria. In case of HGT, newly acquired genes slowly mutate until their statistics are close to the (host) genome average. Unlike them, genes within a bacterial genome do not need to match eukaryotic standards, so the corresponding reads or contigs would be different from the rest of the assembly.
In conclusion, we suggest that limiting contamination control to any single method could possibly mislead the researcher in either direction. A combination of NGS-based amplicon sequencing (ideally from the same DNA samples that would be used for library preparation, to detect any possible contamination in molecular biology equipment or reagents), microscopy-based methods and computational controls of read libraries (if available) is necessary to ensure the axenicity of diatom cultures.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}