
Assessment of the Antimicrobial and Antiproliferative Activities of Chloropyrazine-Tethered Pyrimidine Derivatives: In Vitro, Molecular Docking, and In-Silico Drug-Likeness Studies


Abstract
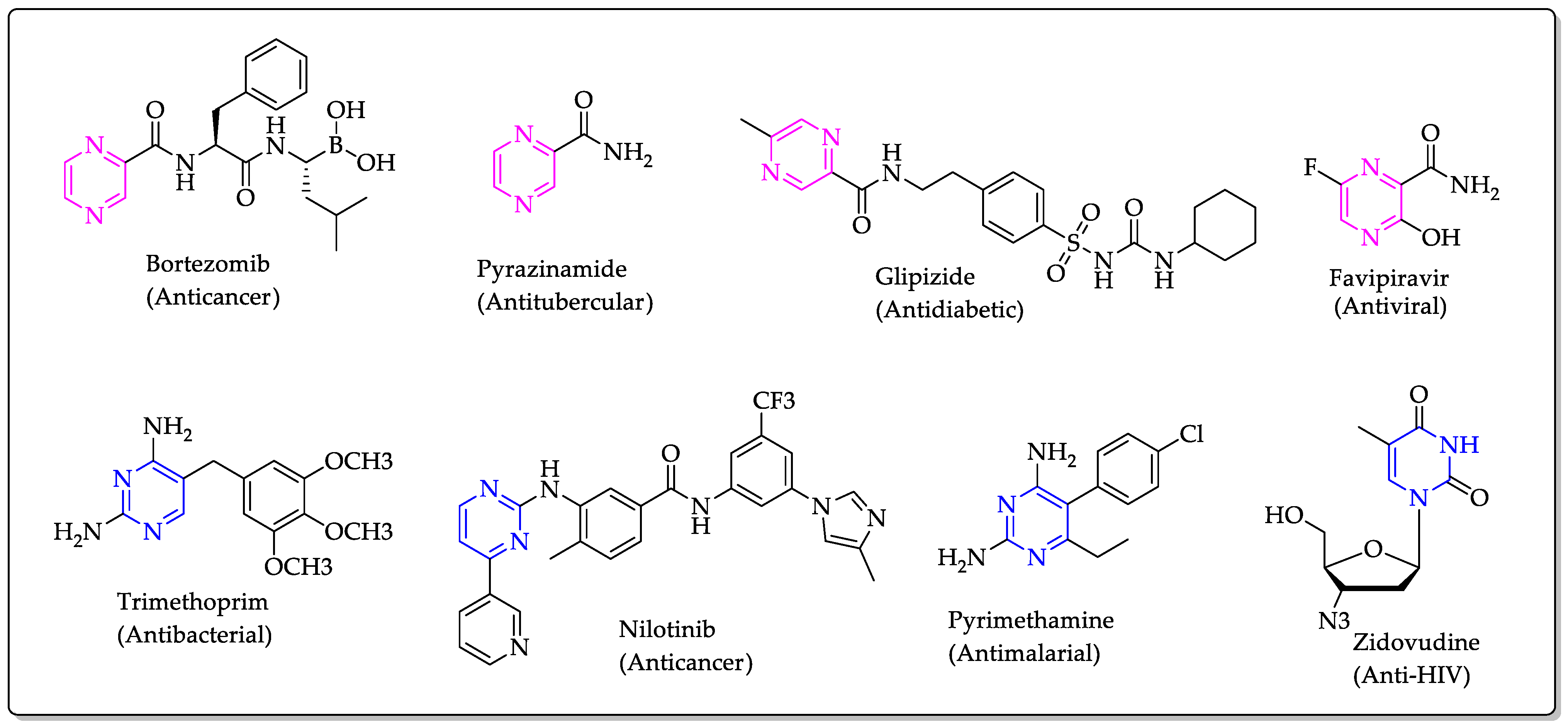
:1. Introduction
2. Results & Discussion
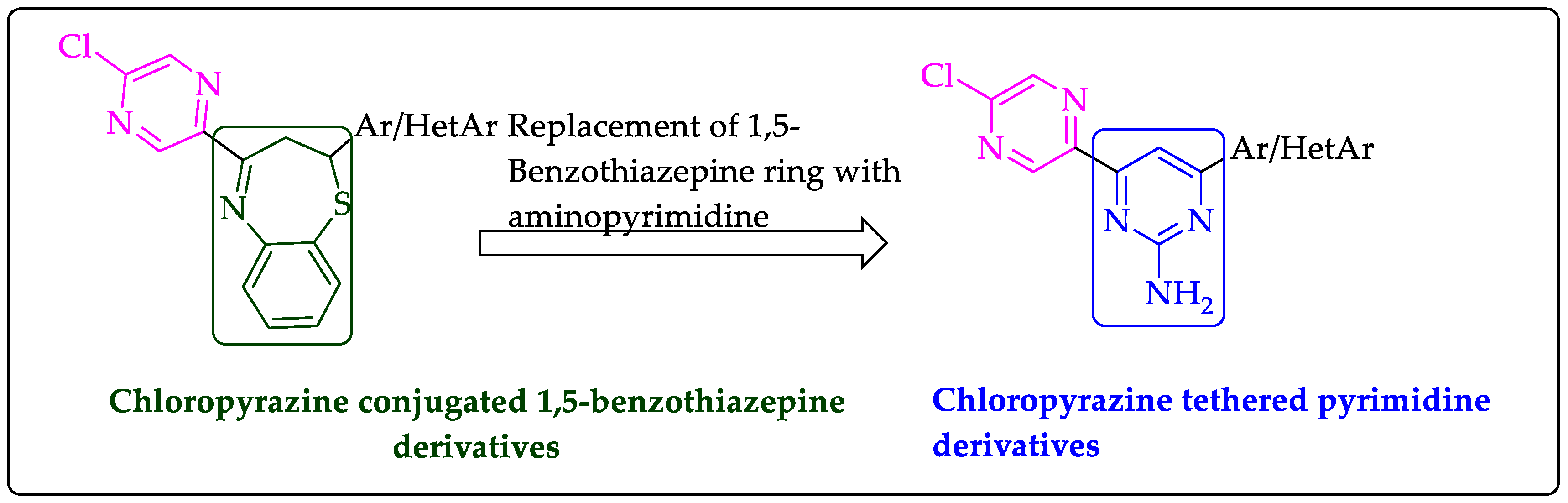
2.1. Chemistry
2.2. Biological Studies
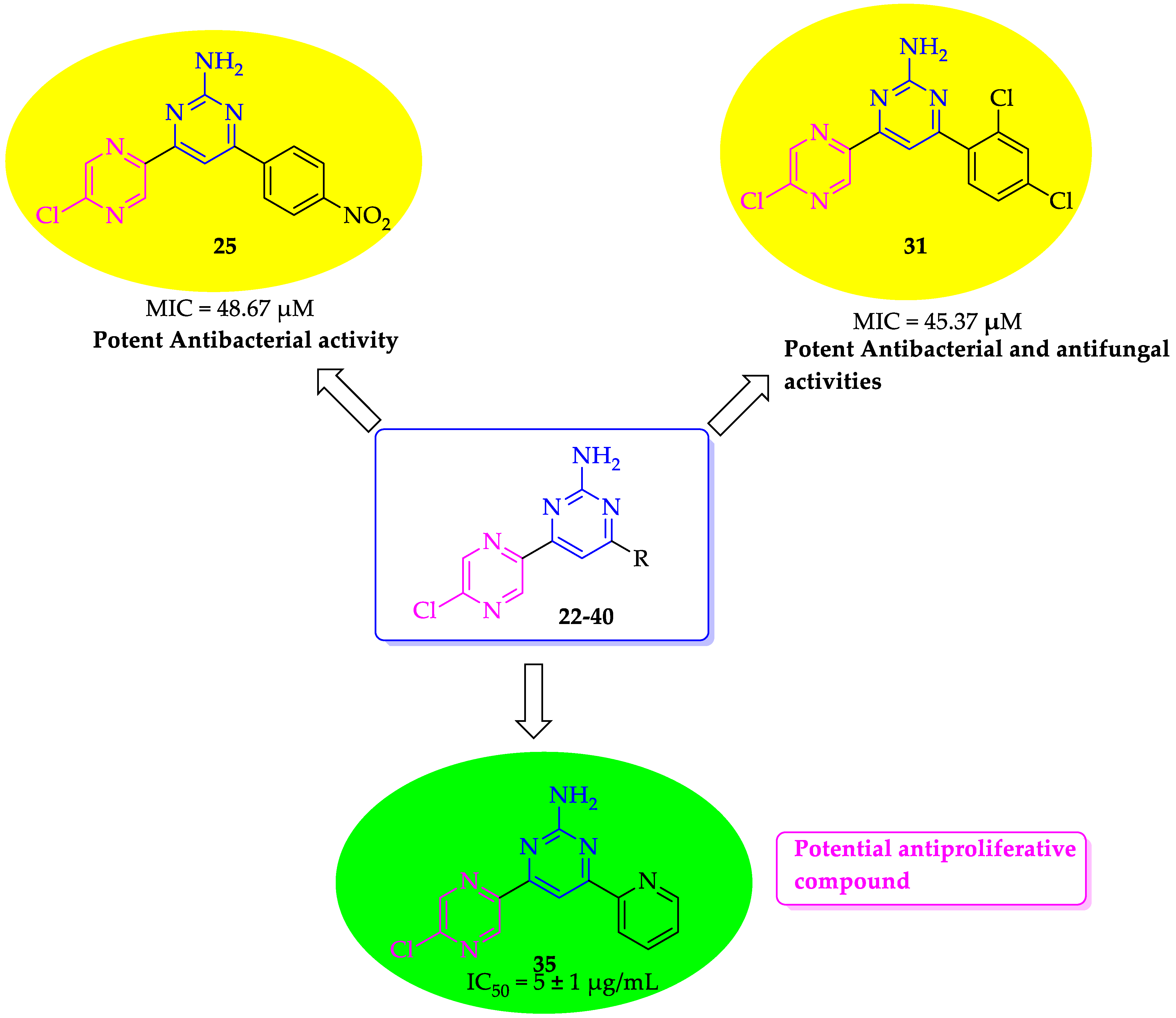
2.2.1. Antibacterial Activity
2.2.2. Antifungal Activity
2.2.3. Antiproliferative Activity
2.3. In Silico Studies
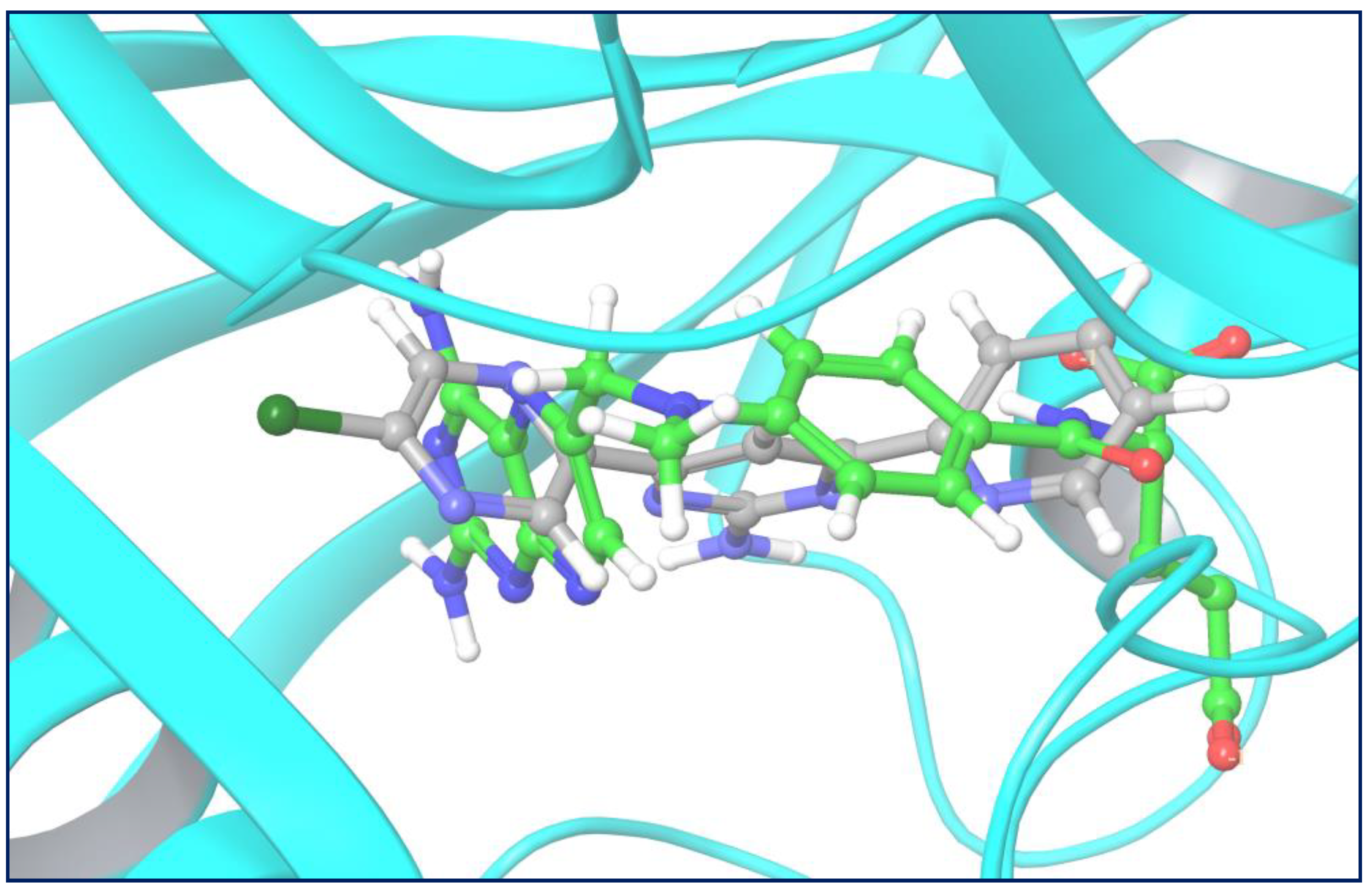
2.3.1. Molecular Docking
2.3.2. In Silico Drug-Likeness Studies
3. Materials and Methods
3.1. Biological Studies
3.1.1. Antibacterial and Antifungal Activities
3.1.2. Antiproliferative Activity
3.2. In Silico Studies
3.2.1. Molecular Docking
3.2.2. In Silico Drug Likeness Prediction
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Gomtsyan, A. Heterocycles in drugs and drug discovery. Chem. Heterocycl. Compd. 2012, 48, 7–10. [Google Scholar] [CrossRef]
- Burch, J.D.; Farand, J.; Colucci, J.; Sturino, C.; Ducharme, Y.; Friesen, R.W.; Lévesque, J.F.; Gagné, S.; Wrona, M.; Therien, A.G.; et al. Naphthalene/quinoline amides and sulfonylureas as potent and selective antagonists of the EP4 receptor. Bioorg. Med. Chem. Lett. 2011, 21, 1041–1046. [Google Scholar] [CrossRef]
- Viegas-Junior, C.; Danuello, A.; da Silva Bolzani, V.; Barreiro, E.J.; Fraga, C.A.M. Molecular hybridization: A useful tool in the design of new drug prototypes. Curr. Med. Chem. 2007, 14, 1829–1852. [Google Scholar] [CrossRef]
- FDA. Novel Drug Approvals for 2021. Available online: https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2021 (accessed on 19 March 2021).
- Miniyar, P.B.; Murumkar, P.R.; Patil, P.S.; Barmade, M.A.; Bothara, K.G. Unequivocal role of pyrazine ring in medicinally important compounds: A review. Mini Rev. Med. Chem. 2013, 13, 1607–1625. [Google Scholar] [CrossRef]
- Semelkova, L.; Konecna, K.; Paterova, P.; Kubicek, V.; Kunes, J.; Novakova, L.; Marek, J.; Naesens, L.; Pesko, M.; Kralova, K.; et al. Synthesis and Biological Evaluation of N-Alkyl-3-(alkylamino)-pyrazine-2-carboxamides. Molecules 2015, 20, 8687–8711. [Google Scholar] [CrossRef]
- Panda, S.S.; Detistov, O.S.; Girgis, A.S.; Mohapatra, P.P.; Samir, A.; Katritzky, A.R. Synthesis and molecular modeling of antimicrobial active fluoroquinolone–pyrazine conjugates with amino acid linkers. Bioorg. Med. Chem. Lett. 2016, 26, 2198–2205. [Google Scholar] [CrossRef] [PubMed]
- Kucerova-Chlupacova, M.; Vyskovska-Tyllova, V.; Richterova-Finkova, L.; Kunes, J.; Buchta, V.; Vejsova, M.; Paterova, P.; Semelkova, L.; Jandourek, O.; Opletalova, V. Novel halogenated pyrazine-based chalcones as potential antimicrobial drugs. Molecules 2016, 21, 1421. [Google Scholar] [CrossRef]
- Semelková, L.; Janďourek, O.; Konečná, K.; Paterová, P.; Navrátilová, L.; Trejtnar, F.; Kubíček, V.; Kuneš, J.; Doležal, M.; Zitko, J. 3-Substituted N-Benzylpyrazine-2-carboxamide Derivatives: Synthesis, Antimycobacterial and Antibacterial Evaluation. Molecules 2017, 22, 495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassin, J.P.; Botha, M.J.; Garikipati, R.; Goyal, M.; Martin, L.; Shah, A. Synthesis and Antibacterial Activity of Benzo [4,5] isothiazolo [2, 3-a] pyrazine-6, 6-dioxide Derivatives. Molecules 2017, 22, 1889. [Google Scholar] [CrossRef] [Green Version]
- Mangrolia, U.; Osborne, W.J. Staphylococcus xylosus VITURAJ10: Pyrrolo[1,2α]pyrazine-1,4-dione,hexahydro-3-(2-methylpropyl)(PPDHMP) producing, potential probiotic strain with antibacterial and anticancer activity. Microb. Pathog. 2020, 147, 104259. [Google Scholar] [CrossRef]
- Kratky, M.; Vinsova, J.; Buchta, V. In vitro antibacterial and antifungal activity of salicylanilide pyrazine-2-carboxylates. Med. Chem. 2012, 8, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Kucerova-Chlupacova, M.; Kunes, J.; Buchta, V.; Vejsova, M.; Opletalova, V. Novel pyrazine analogs of chalcones: Synthesis and evaluation of their antifungal and antimycobacterial activity. Molecules 2015, 20, 1104–1117. [Google Scholar] [CrossRef] [Green Version]
- Zaki, R.M.; Kamal, E.D.; Radwan, S.M.; Abd ul-Malik, M.A. A convenient synthesis, reactions and biological activities of some novel thieno [3, 2-e] pyrazolo [3, 4-b] pyrazine compounds as anti-microbial and anti-inflammatory agents. Curr. Org. Synth. 2018, 15, 863–871. [Google Scholar] [CrossRef]
- Chylewska, A.; Dąbrowska, A.M.; Ramotowska, S.; Maciejewska, N.; Olszewski, M.; Bagiński, M.; Makowski, M. Photosensitive and pH-dependent activity of pyrazine-functionalized carbazole derivative as promising antifungal and imaging agent. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Goel, R.; Luxami, V.; Paul, K. Synthesis, in vitro anticancer activity and SAR studies of arylated imidazo[1,2-a]pyrazine–coumarin hybrids. RSC Adv. 2015, 5, 37887–37895. [Google Scholar] [CrossRef]
- El-Kashef, H.; El-Emary, T.; Verhaeghe, P.; Vanelle, P.; Samy, M. Anticancer and anti-inflammatory activities of some new pyrazolo [3, 4-b] pyrazines. Molecules 2018, 23, 2657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Yuan, X.; Qian, H.; Li, N.; Wang, J. Design, Synthesis, and Biological Evaluation of Two Series of Novel A-Ring Fused Steroidal Pyrazines as Potential Anticancer Agents. Int. J. Mol. Sci. 2020, 21, 1665. [Google Scholar] [CrossRef] [Green Version]
- Seo, Y.; Lee, J.H.; Park, S.H.; Namkung, W.; Kim, I. Expansion of chemical space based on a pyrrolo[1,2-a]pyrazine core: Synthesis and its anticancer activity in prostate cancer and breast cancer cells. Eur. J. Med. Chem. 2020, 188, 111988. [Google Scholar] [CrossRef] [PubMed]
- Raveesha, R.; Kumar, M.G.D.; Prasad, S.B.B. Synthesis of 3-Trifluoromethyl-5, 6-dihydro-[1,2,4] triazolo Pyrazine Derivatives and Their Anti-Cancer Studies. Molbank 2020, 4, M1173. [Google Scholar] [CrossRef]
- Zaki, R.M.; Abdul-Malik, M.A.; Saber, S.H.; Radwan, S.M.; El-Dean, A.M.K. A convenient synthesis, reactions and biological evaluation of novel pyrazolo [3,4-b] selenolo [3,2-e] pyrazine heterocycles as potential anticancer and antimicrobial agents. Med. Chem. Res. 2020, 29, 2130–2145. [Google Scholar] [CrossRef]
- Packiaraj, S.; Kousalya, L.; Pushpaveni, A.; Poornima, S.; Puschmann, H.; Govindarajan, S. Structural and antioxidant properties of guanylhydrazinium pyrazine-2-carboxylate. J. Mater. Sci. Mater. Electron. 2021, 32, 1–15. [Google Scholar] [CrossRef]
- Kucerova-Chlupacova, M.; Dosedel, M.; Kunes, J.; Soltesova-Prnova, M.; Majekova, M.; Stefek, M. Chalcones and their pyrazine analogs: Synthesis, inhibition of aldose reductase, antioxidant activity, and molecular docking study. Monatsh. Chem. 2018, 149, 921–929. [Google Scholar] [CrossRef]
- Stepanić, V.; Matijašić, M.; Horvat, T.; Verbanac, D.; Kučerová-Chlupáčová, M.; Saso, L.; Žarković, N. Antioxidant Activities of Alkyl Substituted Pyrazine Derivatives of Chalcones—In Vitro and In Silico Study. Antioxidants 2019, 8, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moghaddas, S.A.; Hossaini, Z.; Zareyee, D. Green synthesis and investigation of antioxidant ability new pyrazines containing pyrrolo [2, 1-a] isoquinolines derivatives. J. Heterocycl. Chem. 2020, 57, 3856–3867. [Google Scholar] [CrossRef]
- Reddyrajula, R.; Dalimba, U. The bioisosteric modification of pyrazinamide derivatives led to potent antitubercular agents: Synthesis via click approach and molecular docking of pyrazine-1, 2, 3-triazoles. Bioorg. Med. Chem. Lett. 2020, 30, 126846. [Google Scholar] [CrossRef] [PubMed]
- Wati, F.A.; Adyarini, P.U.; Fatmawati, S.; Santoso, M. Synthesis of pyrazinamide analogues and their antitubercular bioactivity. Med. Chem. Res. 2020, 29, 2157–2163. [Google Scholar] [CrossRef]
- Das, R.; Mehta, D.K. Evaluation and Docking Study of Pyrazine Containing 1, 3, 4-Oxadiazoles Clubbed with Substituted Azetidin-2-one: A New Class of Potential Antimicrobial and Antitubercular. Drug Res. 2021, 71, 26–35. [Google Scholar] [CrossRef]
- Mallikarjunaswamy, C.; Mallesha, L.; Bhadregowda, D.G.; Pinto, O. Studies on synthesis of pyrimidine derivatives and their antimicrobial activity. Arab. J. Chem. 2017, 10, S484–S490. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, S.V.; Seijas, J.A.; Vazquez-Tato, M.P.; Sarkate, A.P.; Karnik, K.S.; Nikalje, A.P.G. Ionic liquid-promoted synthesis of novel chromone-pyrimidine coupled derivatives, antimicrobial analysis, enzyme assay, docking study and toxicity study. Molecules 2018, 23, 440. [Google Scholar] [CrossRef] [Green Version]
- Fouda, A.M.; Abbas, H.A.S.; Ahmed, E.H.; Shati, A.A.; Alfaifi, M.Y.; Elbehairi, S.E.I. Synthesis, in vitro antimicrobial and cytotoxic activities of some new pyrazolo[1,5-a]pyrimidine derivatives. Molecules 2019, 24, 1080. [Google Scholar] [CrossRef] [Green Version]
- Sui, Y.F.; Li, D.; Wang, J.; Bheemanaboina, R.R.Y.; Ansari, M.F.; Gan, L.L.; Zhou, C.H. Design and biological evaluation of a novel type of potential multi-targeting antimicrobial sulfanilamide hybrids in combination of pyrimidine and azoles. Bioorg. Med. Chem. Lett. 2020, 30, 126982. [Google Scholar] [CrossRef]
- Naglah, A.M.; Askar, A.A.; Hassan, A.S.; Khatab, T.K.; Al-Omar, M.A.; Bhat, M.A. Biological evaluation and molecular docking with in silico physicochemical, pharmacokinetic and toxicity prediction of pyrazolo[1,5-a]pyrimidines. Molecules 2020, 25, 1431. [Google Scholar] [CrossRef] [Green Version]
- Gomha, S.M.; Zaki, Y.H.; Abdelhamid, A.O. Utility of 3-Acetyl-6-bromo-2H-chromen-2-one for the synthesis of new heterocycles as potential antiproliferative agents. Molecules 2015, 20, 21826–21839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbitskiy, E.V.; Cheprakova, E.M.; Slepukhin, P.A.; Kravchenko, M.A.; Skornyakov, S.N.; Rusinov, G.L.; Chupakhin, O.N.; Charushin, V.N. Synthesis, and structure–activity relationship for C (4) and/or C (5) thienyl substituted pyrimidines, as a new family of antimycobacterial compounds. Eur. J. Med. Chem. 2015, 97, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Singh, L.; Chibale, K.; Singh, K. Structure elaboration of isoniazid: Synthesis, in silico molecular docking and antimycobacterial activity of isoniazid–pyrimidine conjugates. Mol. Divers. 2020, 24, 949–955. [Google Scholar] [CrossRef]
- Santoso, K.T.; Brett, M.W.; Cheung, C.Y.; Cook, G.M.; Stocker, B.L.; Timmer, M.S. Synthesis of FunctionalisedChromonyl-pyrimidines and Their Potential as Antimycobacterial Agents. ChemistrySelect 2020, 5, 4347–4355. [Google Scholar] [CrossRef]
- Dar’in, D.; Rogacheva, E.; Kraeva, L.; Levin, O.; Manicheva, O.; Dogonadze, M.; Vinogradova, T.; Bakulina, O.; Krasavin, M. Mutually Isomeric 2-and 4-(3-nitro-1, 2, 4-triazol-1-yl) pyrimidines Inspired by an Antimycobacterial Screening Hit: Synthesis and Biological Activity against the ESKAPE Panel of Pathogens. Antibiotics 2020, 9, 666. [Google Scholar] [CrossRef]
- Kostova, I.; Atanasov, P.Y. Antioxidant Properties of Pyrimidine and Uracil Derivatives. Curr. Org. Chem. 2017, 21, 2096–2108. [Google Scholar] [CrossRef]
- Emam, D.R.; Alhajoj, A.M.; Elattar, K.M.; Kheder, N.A.; Fadda, A.A. Synthesis and evaluation of curcuminoid analogues as antioxidant and antibacterial agents. Molecules 2017, 22, 971. [Google Scholar] [CrossRef] [Green Version]
- Naidu Kalla, R.M.; Karunakaran, R.S.; Balaji, M.; Kim, I. Catalyst-Free Synthesis of Xanthene and Pyrimidine-Fused Heterocyclic Derivatives at Water-Ethanol Medium and Their Antioxidant Properties. ChemistrySelect 2019, 4, 644–649. [Google Scholar] [CrossRef]
- Abdelgawad, M.A.; Bakr, R.B.; Ahmad, W.; Al-Sanea, M.M.; Elshemy, H.A. New pyrimidine-benzoxazole/benzimidazole hybrids: Synthesis, antioxidant, cytotoxic activity, in vitro cyclooxygenase and phospholipase A2-V inhibition. Bioorg. Chem. 2019, 92, 103218. [Google Scholar] [CrossRef]
- Reddy, O.S.; Suryanarayana, C.V.; Narayana, K.J.P.; Anuradha, V.; Babu, B.H. Synthesis and cytotoxic evaluation for some new 2, 5-disubstituted pyrimidine derivatives for anticancer activity. Med. Chem. Res. 2015, 24, 777–1788. [Google Scholar] [CrossRef]
- Shi, J.B.; Tang, W.J.; Li, R.; Liu, X.H. Novel pyrazole-5-carboxamide and pyrazole–pyrimidine derivatives: Synthesis and anticancer activity. Eur. J. Med. Chem. 2015, 90, 889–896. [Google Scholar] [CrossRef]
- Abdallah, M.A.; Gomha, S.M.; Abbas, I.M.; Kazem, M.S.; Alterary, S.S.; Mabkhot, Y.N. An efficient synthesis of novel pyrazole-based heterocycles as potential antitumor agents. Appl. Sci. 2017, 7, 785. [Google Scholar] [CrossRef] [Green Version]
- Murahari, M.; Prakash, K.V.; Peters, G.J.; Mayur, Y.C. Acridone-pyrimidine hybrids-design, synthesis, cytotoxicity studies in resistant and sensitive cancer cells and molecular docking studies. Eur. J. Med. Chem. 2017, 139, 961–981. [Google Scholar] [CrossRef]
- Abbass, E.M.; Khalil, A.K.; El-Naggar, A.M. Eco-friendly synthesis of novel pyrimidine derivatives as potential anticancer agents. J. Heterocycl. Chem. 2020, 57, 1154–1164. [Google Scholar] [CrossRef]
- Radwan, M.A.; Alminderej, F.M.; Awad, H.M. One-pot multicomponent synthesis and cytotoxic evaluation of novel 7-substituted-5-(1H-indol-3-yl)tetrazolo[1,5-a]pyrimidine-6-carbonitrile. Molecules 2020, 25, 255. [Google Scholar] [CrossRef] [Green Version]
- Madia, V.N.; Nicolai, A.; Messore, A.; De Leo, A.; Ialongo, D.; Tudino, V.; Saccoliti, F.; De Vita, D.; Scipione, L.; Artico, M.; et al. Design, Synthesis and Biological Evaluation of New Pyrimidine Derivatives as Anticancer Agents. Molecules 2021, 26, 771. [Google Scholar] [CrossRef]
- Bai, S.; Liu, S.; Zhu, Y.; Wu, Q. Asymmetric synthesis and antiviral activity of novel chiral amino-pyrimidine derivatives. Tetrahedron. Lett. 2018, 59, 3179–3183. [Google Scholar] [CrossRef]
- Wu, L.; Liu, Y.; Li, Y. Synthesis of spirooxindole-O-naphthoquinone-tetrazolo[1,5-a]pyrimidine hybrids as potential anticancer agents. Molecules 2018, 23, 2330. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Deep, A.; Narasimhan, B. A review on synthesis, anticancer and antiviral potentials of pyrimidine derivatives. Curr. Bioact. Compd. 2019, 15, 289–303. [Google Scholar] [CrossRef]
- Yates, M.K.; Chatterjee, P.; Flint, M.; Arefeayne, Y.; Makuc, D.; Plavec, J.; Spiropoulou, C.F.; Seley-Radtke, K.L. Probing the effects of pyrimidine functional group switches on acyclic fleximer analogues for antiviral activity. Molecules 2019, 24, 3184. [Google Scholar] [CrossRef] [Green Version]
- Shaik, A.B.; Bhandare, R.R.; Nissankararao, S.; Lokesh, B.V.S.; Shahanaaz, S.; Rahman, M.M. Synthesis, and biological screening of chloropyrazine conjugated benzothiazepine derivatives as potential antimicrobial, antitubercular and cytotoxic agents. Arab. J. Chem. 2021, 14, 102915. [Google Scholar] [CrossRef]
- Vegesna, S.; Yejella, R.P.; Shaik, A.B. Synthesis and antitubercular evaluation of some novel pyrimidine derivatives. Int. J. Res. Pharm. Chem. 2017, 7, 181–186. [Google Scholar]
- Raimondi, M.V.; Randazzo, O.; La Franca, M.; Barone, G.; Vignoni, E.; Rossi, D.; Collina, S. DHFR Inhibitors: Reading the Past for Discovering Novel Anticancer Agents. Molecules 2019, 24, 1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger Suite 2014-3; Schrödinger, LLC.: New York, NY, USA, 2014.
- Cody, V.; Luft, J.R.; Pangborn, W. Understanding the role of Leu22 variants in methotrexate resistance: Comparison of wild-type and Leu22Arg variant mouse and human dihydrofolate reductase ternary crystal complexes with methotrexate and NADPH. Acta Crystallogr. D Biol. Crystallogr. 2005, 61, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Vegesna, S.; Yejella, R.P.; Shaik, A.B. Antimicrobial evaluation of some novel pyrazine based chalcones. Int. J. Adv. Pharm. Sci. 2017, 8, 11–18. [Google Scholar]
- Swiss Institute of Bioinformatics. SwissADME. Available online: http://www.swissadme.ch/ (accessed on 17 September 2021).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
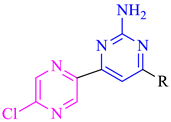
| Compound No. | R | B. subtilis (MIC = µM) | S. aureus (MIC = µM) | E. coli (MIC in µM) | P. aeruginosa (MIC in µM) |
|---|---|---|---|---|---|
| 22 | Phenyl | >200 ± 0.92 | >200 ± 0.88 | >200 ± 0.53 | >200 ± 0.43 |
| 23 | 4″-chlorophenyl | 50.28 ± 0.09 | 50.28 ± 0.28 | 50.28 ± 0.19 | 50.28 ± 0.89 |
| 24 | 4″-fluorophenyl | 53.03 ± 0.38 | 53.03 ± 0.56 | 53.03 ± 0.28 | 53.03 ± 0.62 |
| 25 | 4″-nitrophenyl | 48.67 ± 0.66 | 48.67 ±0.43 | 48.67 ± 0.22 | 48.67 ± 0.42 |
| 26 | 4″-hydroxyphenyl | >200 ± 0.58 | >200 ± 0.15 | >200 ± 0.19 | >200 ± 0.22 |
| 27 | 4″-methylphenyl | >200 ± 0.23 | >200 ± 0.62 | >200 ± 0.38 | >200 ± 0.73 |
| 28 | 4″-methoxyphenyl | >200 ± 091 | >200 ± 0.45 | >200 ± 0.38 | >200 ± 0.56 |
| 29 | 4″-dimethylaminophenyl | 195.84 ± 0.16 | >200 ± 0.92 | >200 ± 0.55 | >200 ± 0.75 |
| 30 | 2″,4″-difluorophenyl | 50.04 ± 0.31 | 50.04 ± 0.21 | 50.04 ± 0.81 | 50.04 ± 0.88 |
| 31 | 2″,4″-dichlorophenyl | 45.37 ± 0.63 | 45.37 ± 0.33 | 45.37 ± 0.29 | 45.37 ± 0.54 |
| 32 | 3″,4″-dimethoxyphenyl | >200 ± 0.47 | >200 ± 0.82 | >200 ± 0.94 | >200 ± 0.18 |
| 33 | 3″-methoxy-4″-hydroxyphenyl | >200 ± 0.52 | >200 ± 0.71 | >200 ± 0.79 | >200 ± 0.95 |
| 34 | 3″,4″,5″-trimethoxyphenyl | 85.60 ± 0.28 | 85.60 ± 0.66 | 85.60 ± 0.56 | 85.60 ± 0.56 |
| 35 | 2″-pyridinyl | >200 ± 0.62 | >200 ± 0.59 | >200 ± 0.22 | >200 ± 0.28 |
| 36 | 3″-pyridinyl | >200 ± 0.93 | >200 ± 0.83 | >200 ± 0.38 | >200 ± 0.33 |
| 37 | 4″-pyridinyl | >200 ± 0.99 | >200 ± 0.77 | >200 ± 0.76 | >200 ± 0.39 |
| 38 | 2″-thienyl | >200 ± 0.91 | >200 ± 0.32 | >200 ± 0.43 | >200 ± 0.53 |
| 39 | 2″-furfuryl | >200 ± 0.82 | >200 ± 0.41 | >200 ± 0.24 | >200 ± 0.77 |
| 40 | 5″-pyrrolyl | >200 ± 0.65 | >200 ± 0.59 | >200 ± 0.61 | >200 ± 0.82 |
| Ciprofloxacin | 145.71 ± 0.18 | 145.71 ± 0.18 | 72.85 ± 0.33 | 72.85 ± 0.33 |
| Compound No. | R | A. niger (MIC = µM ± SD) | C. tropicalis (MIC = µM ± SD) | * DU-145 (IC50 = µg/mL ± SD) | * Human Normal Liver Cells (L02; µg/mL) |
|---|---|---|---|---|---|
| 22 | Phenyl | >200 ± 0.91 | >200 ± 0.58 | 40 ± 1 | >40 |
| 23 | 4″-chlorophenyl | 100.57 ± 0. 52 | 100.57 ± 0.16 | 18 ± 2 | >40 |
| 24 | 4″-fluorophenyl | 106.06 ± 0.19 | 106.06 ± 0.88 | 47 ± 2 | >40 |
| 25 | 4″-nitrophenyl | 97.34 ± 0.28 | 97.34 ± 0.52 | 112 ± 2 | >40 |
| 26 | 4″-hydroxyphenyl | >200 | >200 ± 0.66 | 121 ± 2 | >40 |
| 27 | 4″-methylphenyl | >200 | >200 ± 0.73 | 132 ± 2 | >40 |
| 28 | 4″-methoxyphenyl | >200 | >200 ± 0.22 | 88 ± 2 | >40 |
| 29 | 4″-dimethylaminophenyl | 195.84 ± 0.98 | >200 ± 0.81 | 24 ± 2 | >40 |
| 30 | 2″,4″-difluorophenyl | 50.04 ± 1 | 50.04 ± 0.22 | 10 ± 2 | >40 |
| 31 | 2″,4″-dichlorophenyl | 45.37 ± 0.66 | 45.37 ± 0.18 | 12 ± 2 | >40 |
| 32 | 3″,4″-dimethoxyphenyl | 46.52 ± 0.99 | 186.17 ± 0.63 | 72 ± 2 | >40 |
| 33 | 3″-methoxy-4″-hydroxyphenyl | >200 ± 0.87 | >200 ± 0.29 | 110 ± 1 | >40 |
| 34 | 3″,4″,5″-trimethoxyphenyl | 85.60 ± 0.55 | 85.60 ± 0.90 | 56 ± 2 | >40 |
| 35 | 2″-pyridinyl | 112.39 ± 0.77 | 56.19 ± 0.34 | 5 ± 1 | >40 |
| 36 | 3″-pyridinyl | 112.39 ± 0.23 | 112.39 ± 0.30 | 78 ± 2 | >40 |
| 37 | 4″-pyridinyl | >200 ± 0.58 | 112.39 ± 0.61 | 52 ± 2 | >40 |
| 38 | 2″-thienyl | 110.44 ± 0.71 | 110.44 ± 0.38 | 82 ± 2 | >40 |
| 39 | 2″-furfuryl | 116.92 ± 0.82 | >200 ± 0.72 | 96 ± 2 | >40 |
| 40 | 5″-pyrrolyl | >200 ± 0.85 | >200 ± 0.53 | 105 ± 1 | >40 |
| Fluconazole | - | 84.14 ± 0.19 | 63.10 ± 0.44 | - | - |
| Methotrexate | - | - | - | 11 ± 1 | >75 |
| S. No. | Ligand Name | Docking Score | Interactions | ||
|---|---|---|---|---|---|
| H-Bonds | π–π | Hydrophobic | |||
| 1 | Compound 35 | −6.834 | Glu30, Thr56 | Phe34 | Ile7, Val8, Ala9, Leu22, Phe31, Tyr33, Phe34, Ile60, Leu67, Val115, Tyr121 |
| 2 | Compound 27 | −5.211 | Glu30 | Phe34 | Val8, Ala9, Leu22, Phe34, Ile60, Leu67, Tyr121 |
| 3 | Methotrexate | −11.808 | Ile7, Glu30, Gln35, Arg70, Val115 | - | Ile7, Val8, Ala9, Leu22, Phe31, Tyr33, Phe34, Ile60, Pro61, Leu67, Val115, Tyr121 |
| Compound # | GI Absorption | Lipinski #Violations | CYP2D6 Inhibitor | CYP2C19 Inhibitor |
|---|---|---|---|---|
| 35 | High | 0 | Yes | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhandare, R.R.; Shaik, A.B. Assessment of the Antimicrobial and Antiproliferative Activities of Chloropyrazine-Tethered Pyrimidine Derivatives: In Vitro, Molecular Docking, and In-Silico Drug-Likeness Studies. Appl. Sci. 2021, 11, 10734. https://doi.org/10.3390/app112210734
Bhandare RR, Shaik AB. Assessment of the Antimicrobial and Antiproliferative Activities of Chloropyrazine-Tethered Pyrimidine Derivatives: In Vitro, Molecular Docking, and In-Silico Drug-Likeness Studies. Applied Sciences. 2021; 11(22):10734. https://doi.org/10.3390/app112210734
Chicago/Turabian StyleBhandare, Richie R., and Afzal Basha Shaik. 2021. "Assessment of the Antimicrobial and Antiproliferative Activities of Chloropyrazine-Tethered Pyrimidine Derivatives: In Vitro, Molecular Docking, and In-Silico Drug-Likeness Studies" Applied Sciences 11, no. 22: 10734. https://doi.org/10.3390/app112210734