Abstract
Under the density functional theory framework, we have calculated the electronic and elastic properties of APoO (A = Be, Mg, Ca, Sr, Ba, and Ra) cubic perovskites. We found that CaPoO, SrPoO, BaPoO, and RaPoO are topological insulators (TIs) with very large bandgaps of 0.861, 0.871, 0.820, and 0.810 eV, respectively. The nontrivial band topology together with the Z topological number of APoO perovskite are investigated. We also theoretically determine the three independent elastic constants C, C, and C of the APoO perovskite. The bulk modulus, shear modulus, Young’s modulus, Poisson’s ratio, and anisotropy factor are also calculated from the obtained elastic constants. We found that the Debye temperature for the APoO perovskite is around 330-370 K. In the bulk APoO perovskite, if the center Po atom is shifted 0.09Å away from the center, the induced electric polarization is quite large, being around 0.02 C/m. In the surface band calculation, we found that both AO and PoO surfaces give rise to contributions to the conduction channel. If the Po atom moves both in-plane and out-of-plane, we show that both electric polarization and topologically protect surface conduction states exist in APoO perovskite, indicating that these oxide APoO perovskites are ferroelectric TIs and might be useful for spintronic applications.
1. Introduction
Topological insulators [1,2], which are characterized by an insulating bulk state and a unique protected gapless surface state, have been of particular interest in the past decades. Topological insulators are a new kind of material that have attracted much attention due to their interesting properties and great potential for spintronic devices [3,4]. For example, the metallic surface states of the TIs are linearly dispersed in the momentum space with the helical spin textures that are protected by time-reversal symmetry and are also characterized by the so-called topological invariant [5]. These interesting features have increased the interest in TIs for scientists in both experimental and theoretical [6] research fields. In 2006 [7], the first 3D topological insulator phase was theoretically predicted. The element with a large spin-orbit coupling (SOC) effect is crucial for the topology. Theoretically, not only TI phase itself is interesting, but the combination with other physical properties or phases, such as superconductivity and magnetism [8,9], is also promising.
Several kinds of crystal structures are found or predicted to have a topological insulator phase: for example, BiTe [1] and BiSe [1] in the hexagonal structure; LaPtBi [10,11] half-Heusler compound [9,12,13,14,15,16,17], oxide perovskite [18,19], halide perovskite [20,21,22], binary compound [23], BaBiIO [24], and CaTePoO [25] double perovskites in the cubic structure. The bulk bandgap for these Tls are in general smaller than 0.5 eV; for example, bismuth-based compounds such as BiTe [26] are 0.17 eV, BiTeS [27] 0.3 eV, and PbBiTeS is 0.2 eV in the experiment and 0.3 eV in theory. In 2012, oxide perovskite YBiO [18,19] was also theoretically predicted to be a topological insulator with a bandgap of 0.33 eV. Further investigating cubic perovskites, CsPbI [20,28], CsSnI [29], CsSbBr [29], and CsSnCl [29] were also found to be topological insulators. The bandgap depends on the external pressure. Large bandgap topological insulators are also found in the double perovskites. For example, the bandgaps for BaBiIO [24], SrBiIO [24], CaBiIO [24], BaBiBrO [24], SrBiBrO [24], and CaBiBrO [24] are 0.55, 0.53, 0.52, 0.33, 0.32, and 0.34 eV, respectively. CaTePoO [25], SrTePoO [25], and BaTePoO [25] double perovskites are also predicted to have theoretically large bandgap topological insulators, with bandgaps of 0.325, 0.400, and 0.375 eV, respectively.
Nonetheless, large bandgap Tls could merely be found in two-dimensional structures. For example, PbXF [30], PbC [31], and BiC [31] (X = H, Cl, F, Br, and I) nanoribbons are theoretically predicted to have a dramatically large bandgap ranging between 0.79 eV and 0.99 eV; Bi (111) monolayer [32] on BiTe also has a bandgap of 0.5 eV; PbAF (A = C, Si, Ge, and Sn) is also theoretically predicted to have an indirect bandgap ranging from 0.3 eV to 0.7 eV. Most recently, the PbCB [33] (B = H, F, and Cl) monolayer is also theoretically reported as TI, and the bandgap ranges from 0.84 to 0.98 eV.
Polonium is a material first isolated by Madame Curie. It is a metal and has the lowest electronegativity in the group of chalcogens [34]. Polonium usually shows +2 and +4 oxidation states in their compounds with oxygen [35]. Compared with selenium and tellurium, the magnitude of SOC from polonium is larger and expected to play an important role in the electronic properties of ABO with perovskite structures.
This work aimed to provide a detailed characterization of the electronic structures of the large bandgap oxide APoO perovskites. This paper is organized as follows. The crystal structure and computational method are described in Section 2. The structural and electronic properties without/with spin-orbit coupling taken into account are discussed in Section 3. Further, mechanical and thermodynamic properties are then discussed. In Section 3.3, we display the supercell band structure without and with the surface being electrically polarized. Finally, we make conclusions.
2. Computational Method
The ideal APoO (A = Be, Mg, Ca, Sr, Ba, and Ra) perovskite is a cubic structure with point group 221 ( symmetric) as shown in Figure 1a. The Wyckoff position for the A atom is set at the corner (0, 0, 0), Po atom is set at the center (1/2, 1/2, 1/2), and oxygen atoms are face-centered (1/2, 1/2, 0), (1/2, 0, 1/2), and (0, 1/2, 1/2), respectively. The structural and electronic properties of APoO perovskites are calculated by using the density functional theory as implemented in the Vienna ab initio simulation package (VASP) [36,37] with the generalized gradient approximation (GGA) [38,39] for the exchange-correlation function. The -centered Monkhorst–Pack scheme with a k-mesh of 12 × 12 × 12 is used for the Brillouin zone integration. The self-consistent total energy criterion is set to be 1.0 × 10 eV. In all cases, a plane wave basis set with cut-off energy of 500 eV was used. Equilibrium lattice constant, a, for APoO perovskites is obtained from the structure optimization technique. This equilibrium lattice constant is also compared with the minimum in the calculated total energy as a function of the lattice constant, and the differences are negligible. For example, the equilibrium lattice constant for the BaPoO is 4.482 (Å), and the k-mesh and cut-off energy are identical to the corresponding ones from the structure optimization technique.
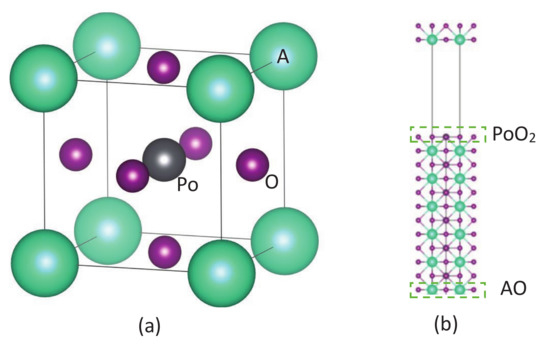
Figure 1.
(a) Crystal structure of APoO (A = Be, Mg, Ca, Sr, Ba, and Ra). (b) Supercell structure contains six formula units with vacuum for the surface band calculation. In a normal supercell, there are PoO and AO surfaces on opposite sides. To investigate which surfaces give rise to the contributions on conduction bands, we prepared two kinds of supercells. One has PoO surface on both sides, i.e., bottom AO layer is removed, and the other has AO surfaces on both sides, i.e., remove the top PoO layer.
Theoretically, one way to classify the topological insulator’s characteristic is the band inversion in momentum space. As suggested by Fu [40], the topological number is calculated by analyzing the parities of the occupied bulk band structure at some specified time reversal invariant momenta in the Brillouin zone. In three dimensions, there are eight time reversal invariant points which lead to four independent invariants (, , , and ). When , the material is a topological insulator. Once the material is predicted to be a TI, the direct results of the surface metallic bands by using a large supercell with vacuum are also important. We used a supercell that contained six formula units of APoO with a vacuum of 20 (Å), as shown in Figure 1b. In this calculation, the k-mesh is 6×6×1, and the energy cut-off is 500 eV.
3. Results and Discussion
3.1. Structural and Electronic Band Structures
Calculated equilibrium lattice constant a (Å), bandgap (eV), and cohesive energy (eV) for the APoO perovskite are listed in Table 1. The cohesive energy is the energy differences between bulk phase and its isolated atoms, i.e., . Calculated cohesive energies were 16.73, 18.76, 22.43, 22.96, 23.70, and 23.03 eV for BePoO, MgPoO, CaPoO, SrPoO, BaPoO, and RaPoO, respectively. The equilibrium lattice constants increased monotonically with the atomic number. We also demonstrate the electronic band structures without and with the spin-orbit coupling effect in Figure 2. Without the spin-orbit coupling effect, we found that all APoO perovskites were ordinary metals. The topology can not be verified merely from the band structure with the SOC effect. It is worth to see how SOC affects the APoO perovskite.

Table 1.
Calculated equilibrium lattice constant a, fully relativistic bandgap and cohesive energy for APoO perovskite.
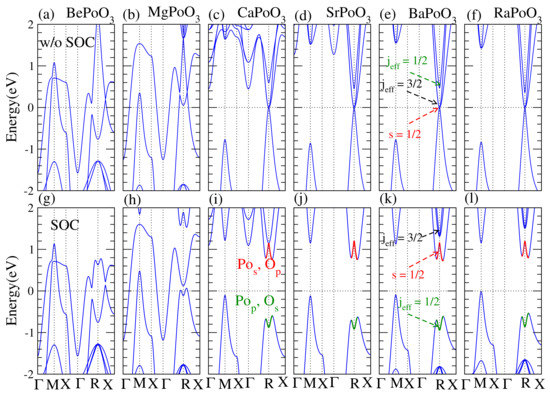
Figure 2.
Scalar relativistic band structures, (a–f) upper panel, and fully relativistic band structures, (g–l) bottom panel for APoO perovskite. The Fermi level is shifted to zero as the reference. Without SOC effect, (a) BePoO, (b) MgPoO, (c) CaPoO, (d) SrPoO, (e) BaPoO, and (f) RaPoO are metals while there are Dirac corn-like band appeared at R point. The highest occupied band at R point is singlet s = 1/2 and the lowest two bands are j = 3/2 and 1/2. When the SOC effect is included, (g) BePoO and (h) MgPoO are metals, however (i) CaPoO, (j) SrPoO, (k) BaPoO, and (l) RaPoO are insulators. The metal to insulator transition appears due to the SOC effect and the s = 1/2 and j = 1/2 bands flipped, see (e) and (k).
We found that CaPoO, SrPoO, BaPoO, and RaPoO were possible topological insulators. There were clear Dirac corn-like bands at R points for the CaPoO, SrPoO, BaPoO, and RaPoO perovskites, see Figure 2 upper panel. When the SOC effect was taken into account, BePoO and MgPoO were still metals, see Figure 2g,h. However, CaPoO, SrPoO, BaPoO, and RaPoO were insulators, and the band was inverted at R point, indicating these perovskites are possible topological insulators.
In previous studies of the YBiO perovskite [18,19,41], the topological phase appeared at equilibrium. Halide perovskites such as CsPbI and CsPbBr can transform into TI under very high pressure. To investigate how the bandgap varies with lattice constants, plotted in Figure 3 is the bandgap as a function of its lattice constants, a. The calculated bandgap was the largest at the equilibrium lattice constant for CaPoO and SrPoO, and the bandgap decreases away from the equilibrium lattice constant. For BaPoO and RaPoO, the largest bandgap occurred at smaller lattice constants. We found that the TI phase existed in a wide range of lattice constants, see Figure 4.
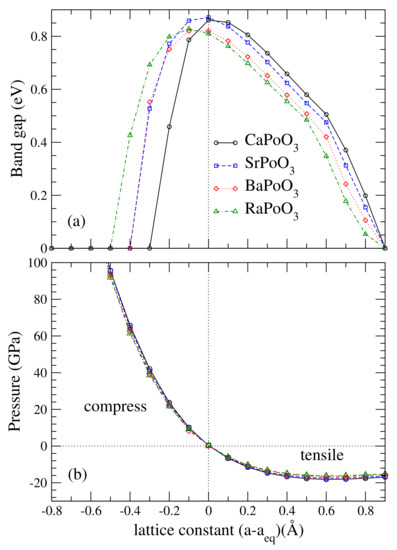
Figure 3.
Calculated (a) fully relativistic band gap, in eV, and (b) external pressure (GPa) as a function of the lattice constant with respect to its equilibrium lattice constant (Å).
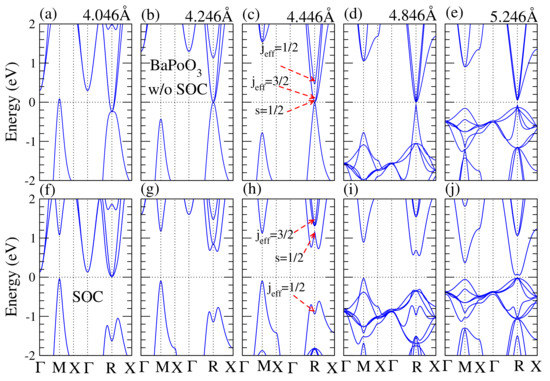
Figure 4.
Calculated electronic band structures of BaPoO as a function of the lattice constant without (upper panel) and with (bottom panel) SOC. Lattice constant for (a,f) is 4.046 Å; (b,g) 4.246 Å; (c,h) is 4.446 Å; (d,i) is 4.846 Å; (e,j) is 5.246 Å. The external pressure induced by the change of lattice constant is from 60 GPa to −20 GPa as shown in Figure 3b. The Fermi level is shifted to zero as a reference. Without SOC effect, the BaPoO perovskite is a normal metal and the band contributions at R point near the Fermi level is the same, except (e). When the SOC is taken into account, the band inversion occurs at lattice constant from 4.046 Å to 4.846 Å. The energy for the lowest unoccupied band is decreased when the lattice constant is decreased or increased, which results in a decrease in bandgap.
To understand how the bandgap varies with lattice constant, we demonstrated the electronic band structure of BaPoO as a function of lattice constant with and without the SOC effect in Figure 4. In the absence of SOC effect, BaPoO was metal until the lattice constance increased to 4.864 (Å). When the SOC effect was taken into account, see Figure 4h, the indirect bandgap came from the energy differences between R and M points. The lowest energy of conduction band near R point decreased as the lattice constant increased, resulting in a decrease in bandgap. In the cubic BaPoO perovskites, the Ba d bands were split into two-fold and three-fold orbitals under cubic symmetry. At point the topmost valence bands were composed of Ba, Po, and O orbitals. The lowest conduction bands at point were composed of Ba and O orbitals. In other words, the energy for the Ba orbitals is higher than the Ba orbitals. Due to Ba and Po hybridization, the Ba bands were located below the Ba bands at the R points. At R point at the Fermi level, the band compositions were in triplets with Ba states, Po states, and O states. Near R point, the valence bands and conduction bands were both composed of Ba states, Po states, and O and O states. These band compositions were very different from the corresponding one at the point, where the topmost valence bands were composed of Ba, Po, and O orbitals, and the lowest conduction bands were composed of Ba and O orbitals.
When the SOC effect was taken into account, we found that at the equilibrium lattice constant, BePoO and MgPoO were still metals. At the same time, CaPoO, SrPoO, BaPoO, and RaPoO became insulators, and the corresponding indirect bandgaps were 0.861, 0.871, 0.820, and 0.810 (eV), respectively. Calculated bandgap is a very large TI bandgap in oxide perovskites. Especially for the PBE result in the DFT calculation usually underestimates the bandgap [29]. The band inversion induced by the SOC effect evidences the topology. To confirm, we also calculated the Z topological numbers. The index is defined as (,) as reported by Fu et al. [7]. Our calculated Z index for the APoO perovskites was (1000), which suggests a strongly topological insulator.
It is fascinating if the cubic perovskites are electrically polarized. Nonetheless, the ideal cubic perovskite has a space inversion structure. Hence the electric polarization, , for all cubic perovskites is zero. However, there were kinds of defects or structural distortions in experiments. These defects or distortions will break the space inversion of the cubic perovskite, which produces a nonzero electric polarization. One way to study the electric polarization is to calculate the induced as a function of the atom’s displacement. Plotted in Figure 5 is the electric polarization as a function of the Po atom’s displacement. This displacement is with respect to its equilibrium lattice constant. For example, the Po atom’s displacements of 0.02 for the CaPoO, SrPoO, BaPoO, and RaPoO were 0.087 (Å), 0.088 (Å), 0.089 (Å), and 0.089 (Å), respectively. The induced electric polarization was large and increased linearly to around 0.02 (C/m) at 0.02 of the Po atom’s displacement. The increased slowly and saturated after the Po atom’s displacement was higher than 0.02. We also found that the calculated bandgap decreased as the Po atom moved away from the center at the TI phase. This behavior was also reported in previous study of the halide CsPbI perovskites [20].
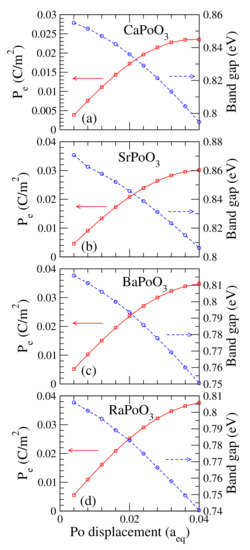
Figure 5.
Calculated electric polarization P, in C/m, and fully relativistic bandgap, in eV, for the (a) CaPoO, (b) SrPoO, (c) BaPoO and (d) RaPoO as a function of the lattice constant with respect to its equilibrium lattice constant (Å).
3.2. Mechanical Properties
The mechanical stability and the related physical properties of the APoO perovskites are listed in Table 2. Due to the cubic symmetry of the perovskite structure, only three independent elastic constants C, C, and C need to be taken into account. These elastic constants are determined from the stress–strain relations, where some finite distortions of the lattice are calculated. In these calculations, we used a k-mesh of 14 × 14 × 14, and the self-consistent total energy criterion was set to be 1.0 × eV. Listed in Table 2 are the elastic constants C, C, and C of the APoO perovskites together with the bulk modulus, B, Voigt shear modulus, , Reuss shear modulus, , shear modulus, G, Young’s modulus, E, anisotropy factor, , Poisson ratio, , and Pugh’s ratio, . These physical properties are calculated from the following equations:
Table 2.
Calculated elastic constants (GPa), (GPa), (GPa), bulk modulus B (GPa), shear modulus G (GPa), Young’s modulus E (GPa), anisotropy factor (arb. unit), Poisson’s ratio (arb. unit), and Pugh’s ratio B/G (arb. unit) of the APoO perovskite.
The mechanical stability criteria [42] for the cubic structure are , , and . We found that the calculated for the APoO perovskites was positive and ranged from 220 GPa to 260 GPa. Similarly, was positive and ranged between 47 GPa and 61 GPa. In all APoO perovskites, the criterion was satisfied, and thus the anisotropy factor was positive. If the anisotropy factor is negative in the cubic structure, which indicates the criterion is not satisfied, the structure is unstable. The criterion can be regarded as an elastic property under the orthogonal distortion with a fixed volume. We notice that the largest of 49.6 GPa in RaPoO was two times larger than the corresponding one in BePoO. However, all were positive. The bulk modulus describes a measure of resistance to the volume change under external pressure. Our calculated bulk modulus was positive and ranged from 110 GPa to 125 GPa. This bulk modulus was larger, which implies a strong resistance to the volume change under pressure. We conclude that all stability criteria are satisfied, indicating that APoO perovskites are mechanically stable.
Besides, the stiffness is also an important material qualtiy. We present Young’s modulus E in Table 2. If E is higher, it implies the material is stiffer. The E increased monotonically from 103.5 GPa in BePoO to 155.5 GPa in RaPoO. The calculated Young’s modulus was large, being over 100 GPa.
To understand the bonding characteristics in a material, we calculated the Poisson’s ratio and listed it in Table 2. The Poisson’s ratio describes the expansion or contraction in the direction perpendicular to the external force’s direction. It has the value . It is hard to find a material with a negative Poisson ratio in nature, though it is possible in mathematics. However, there are still experimental suggestions to synthesize these materials [43,44]. Therefore, was between 0 and 0.5. It is shown that the lower limit of was 0.25 for the central force solid, and was the upper limit of the infinite elastic anisotropy. In other words, typical isotropic materials have a Poisson’s ratio of . The Poisson’s ratio is 0.33 for copper and 0.5 for rubber. The present Poisson’s ratio ranged between 0.28 and 0.34, which suggests these materials are intermetallic compounds.
Another physical quantity, B/G, describes the brittle or ductile behavior of materials as suggested by Pugh and known as Pugh’s ratio. The critical value for the B/G ratio is 1.75. A high B/G ratio is associated with ductility, whereas a low value corresponds to brittleness. At zero pressure, our calculated results for all the APoO perovskites can be classified as ductile materials since all B/G ratios were larger than 1.75. The Poisson’s ratio can also be used to classify the brittleness and ductility as and , respectively. Moreover, the so-called Cauchy parameter also determines the brittleness and ductility of a material. The Cauchy relation’s judgement is simple; the material is ductile if the Cauchy parameter is positive; otherwise, the material is brittle. Our calculated Cauchy parameters were all positive, which implies the ductile behavior of APoO perovskites.
The Zener anisotropy factor can be used to predict whether the material is isotropic or anisotropic. It takes the value of 1 for isotropy material. As shown in Table 2, the calculated anisotropy was larger than one, indicating that APoO perovskites are elastically anisotropic.
Corresponding to the Debye theory, the Debye temperature of a material correlates to the elastic properties and the thermodynamic properties. The Debye model assumes that the solid is elastic continuum in which all sound waves travel at the same velocity independent of their wavelength. Indeed, is the temperature of a crystal’s highest normal mode of vibration. The lattice vibrations also determine the thermodynamic properties such as thermal expansion and specific heat of a solid. Besides, it also corresponds to the structural stability and the strength of bonds. The Debye temperature can be estimated from the average elastic wave velocity:
where h is the Planck constant, is the Boltzmann constant, n is the number of atoms per formula, V is the atomic volume, and is the average sound velocity.
To obtain average sound velocity , we first calculate the longitudinal, , and transverse, , sound velocity from Navier’s equation:
and
Calculated , , , and values are listed in Table 3; indeed, there are no experimental and theoretical results in APoO perovskite. The Debye temperatures for most crystals were around 200 to 400 Kelvin.
Table 3.
Calculated density (), the longitudinal, (m/s), transverse, (m/s), average sound velocity, (m/s), and Debye temperature (K) from the elastic moduli.
3.3. Surface Band Structures
The surface conduction bands for the topological insulators are the most essential properties because these metallic surface states allow electrons to travel insensitive to scattering by impurities. In other words, these metallic surface states are useful for spintronic applications. To investigate the behavior of the surface conduction bands, we prepared a supercell containing 12 layers of APoO perovskites (six formula units) with a vacuum of 20 (Å). Plotted in Figure 6 is the band structures of the SrPoO slab. Figure 6a shows the supercell band structure calculations. In this calculation, the supercell contains six formula units of SrPoO with SrO and PoO surfaces on each side. There were clear conduction bands near M points, indicating that SrPoO is a conductor. These conduction bands are topologically protected. It is interesting to see where these surface conduction bands come from. To verify, we prepared two kinds of supercells: one contains the SrO surface on both sides, denoted as SrO plane, and the other includes the PoO surface on both sides, denoted as PoO plane. The band structures are shown in Figure 6b. We found that both SrO and PoO surfaces gave rise to the contributions of the conduction bands; the red-dashed bands come from the SrO surface while green solid bands come from the PoO surface.
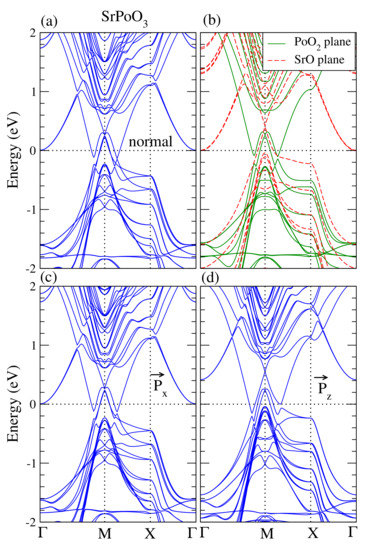
Figure 6.
(a) Calculated fully relativistic band structure for the SrPoO supercell. (b) Fully relativistic band structures with PoO surfaces on both sides, green solid-lines, and SrO surfaces on both sides, red-dashed lines. (c,d) Fully relativistic band structures with Po atoms moving in-plane and out-of-plane, respectively.
We studied the electric polarization if the Po atom is moved away from the center in bulk form. However, it is interesting to see if the surface also being electrically polarized. In the surface calculations, the electric polarization has two kinds of freedom: one is the electric polarization produced by the displacement of the Po atom along the x-direction, in-plane, and the other is along the z-direction, out-of-plane. Plotted in Figure 6c is the calculated surface band structures with electric polarization that is in-plane. This result was obtained with the Po atom moving in the x-direction about 0.02 (Å). The result was almost identical to the corresponding one in the normal supercell calculations. If the Po atom moves out-of-plane about 0.02 (Å) which produces an electric polarization, see Figure 6d, the topologically protected surface conduction bands still exist.
4. Conclusions
We studied the electronic and mechanical properties of oxide APoO (A = Be, Mg, Ca, Sr, Ba, and Ra) cubic perovskite by using density functional theory. We predict that CaPoO, SrPoO, BaPoO, and RaPoO are topological insulators with a very large bandgaps of 0.861, 0.871, 0.820, and 0.810 eV, respectively. This bandgap is the largest among the present reported 3D topological insulators. The nontrivial band topology together with the Z topological number of APoO perovskite is also studied. We also found that all APoO perovskites are mechanically stable. The Pugh’s ratio is larger than 1.75, indicating these APoO perovskites are ductile materials, and the Debye temperature is around 350K. Following the previous study in lead halide CsPbI perovskite, we found the APoO is a topological insulator with the surface being electrically polarized. It suggests a ferroelectric topological insulator in oxide APoO perovskite and might be useful for future spintronic devices.
Author Contributions
Conceptualization, J.-C.T.; methodology, C.-H.L.; formal analysis, J.-C.T., C.-H.L.; writing—original draft preparation, C.-H.L. and J.-C.T.; writing—review and editing, C.-H.L. and J.-C.T.; supervision, J.-C.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Ministry of Science and Technology (MOST), Taiwan, grant numbers 107-2112-M-039-002-MY2 and also funded by the China Medical University(CMU) grant numbers CMU109-N-24.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank the Academia Sinica of the ROC and the NCHC of Taiwan for providing CPU time.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhang, H.; Liu, C.X.; Qi, X.L.; Dai, X.; Fang, Z.; Zhang, S.C. Topological insulators in Bi2Se3, Bi2Te3 and Sb2Te3 with a single Dirac cone on the surface. Nat. Phys. 2009, 5, 438. [Google Scholar] [CrossRef]
- Thouless, D.J.; Kohmoto, M.; Nightingale, M.P.; den Nijs, M. Quantized Hall Conductance in a Two-Dimensional Periodic Potential. Phys. Rev. Lett. 1982, 49, 405. [Google Scholar] [CrossRef]
- Moore, J.E. The birth of topological insulators. Nature 2010, 464, 194. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Wen, J.; Zhou, J. First-principles calculation of Z2 topological invariants within the FP-LAPW formalism. Comput. Phys. Commun. 2012, 183, 1849. [Google Scholar] [CrossRef]
- Deng, M.-X.; Ma, R.; Luo, W.; Shen, R.; Sheng, L.; Xing, D.Y. Time-reversal invariant resonant backscattering on a topological insulator surface driven by a time-periodic gate voltage. Sci. Rep. 2018, 8, 12338. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.-G. Topological orders and edge excitations in fractional quantum Hall states. Adv. Phys. 1995, 44, 405. [Google Scholar] [CrossRef]
- Fu, L.; Kane, C.L.; Mele, E.J. Topological Insulators in Three Dimensions. Phys. Rev. Lett. 2007, 98, 106803. [Google Scholar] [CrossRef]
- Kou, X.; Fan, Y.; Lang, M.; Upadhyaya, P.; Wang, K.L. Magnetic topological insulators and quantum anomalous hall effect. Solid State Commun. 2015, 215–216, 34. [Google Scholar] [CrossRef]
- Li, Z.; Su, H.; Yang, X.; Zhang, J. Electronic structure of the antiferromagnetic topological insulator candidate GdBiPt. Phys. Rev. B 2015, 91, 235128(R). [Google Scholar] [CrossRef]
- Hallouche, A.; Hamri, A.; Kacimi, S.; Zaoui, A. Magnetic ordering in RPtBi topological insulators from DFT+U calculations. Phys. B 2014, 442, 100. [Google Scholar] [CrossRef]
- Li, C.; Wen, Z. Electronic structure of topological insulators with MM’X half-Heusler compounds using density functional theory. Thin Solid Films 2013, 546, 436. [Google Scholar] [CrossRef]
- Feng, W.; Xiao, D.; Zhang, Y.; Yao, Y. Half-Heusler topological insulators: A first-principles study with the Tran-Blaha modified Becke-Johnson density functional. Phys. Rev. B 2010, 82, 235121. [Google Scholar] [CrossRef]
- Lin, H.; Wray, L.A.; Xia, Y.; Xu, S.; Jia, S.; Cava, R.J.; Bansil, A.; Hasan, M.Z. Half-Heusler ternary compounds as new multifunctional experimental platforms for topological quantum phenomena. Nat. Mater. 2010, 7, 546. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.M.; Xu, G.Z.; Du, Y.; Liu, E.K.; Liu, Z.Y.; Wang, W.H.; Wu, G.H. A new class of topological insulators from I-III-IV half-Heusler compounds with strong band inversion strength. J. Appl. Phys. 2014, 115, 083704. [Google Scholar] [CrossRef]
- Zhang, X.M.; Xu, G.Z.; Liu, E.K.; Liu, Z.Y.; Wang, W.H.; Wu, G.H. On the influence of tetrahedral covalent-hybridization on electronic band structure of topological insulators from first principles. J. Appl. Phys. 2015, 117, 045706. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Chen, M.; Yang, X.-B.; Zhao, Y.-J. Theoretical search for half-Heusler topological insulators. Phys. Rev. B 2015, 91, 094107. [Google Scholar] [CrossRef]
- Chadov, S.; Qi, X.; Kübler, J.; Fecher, G.H.; Felser, C.; Zhang, S.C. Tunable multifunctional topological insulators in ternary Heusler compounds. Nat. Mater. 2010, 9, 541. [Google Scholar] [CrossRef]
- Trimarchi, G.; Zhang, X.; Freeman, A.J.; Zunger, A. Structurally unstable AIIIBiO3 perovskites are predicted to be topological insulators but their stable structural forms are trivial band insulators. Phys. Rev. B 2014, 90, 161111(R). [Google Scholar] [CrossRef]
- Jin, H.; Im, J.; Freeman, A.J. Topological insulator phase in halide perovskite structures. Phys. Rev. B 2012, 86, 121102(R). [Google Scholar] [CrossRef]
- Liu, S.; Kim, Y.; Tan, L.Z.; Rappe, A.M. Strain-Induced Ferroelectric Topological Insulator. Nano Lett. 2016, 16, 1663. [Google Scholar] [CrossRef]
- Lang, L.; Yang, J.-H.; Liu, H.-R.; Xiang, H.J.; Gong, X.G. First-principles study on the electronic and optical properties of cubic ABX3 halide perovskites. Phys. Lett. A 2014, 378, 290. [Google Scholar] [CrossRef]
- Weeks, C.; Franz, M. Topological insulators on the Lieb and perovskite lattices. Phys. Rev. B 2010, 82, 085310. [Google Scholar] [CrossRef]
- Bernevig, B.A.; Hughes, T.L.; Chang, S.C. Quantum Spin Hall Effect and Topological Phase Transition in HgTe Quantum Wells. Science 2006, 314, 1757. [Google Scholar] [CrossRef] [PubMed]
- Pi, S.-T.; Wang, H.; Kim, J.; Wu, R.; Wang, Y.-K.; Lu, C.-K. New Class of 3D Topological Insulator in Double Perovskite. J. Phys. Chem. Lett. 2017, 8, 332. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.-H.; Zhou, J.; Pi, S.-T.; Wang, Y.-K. Topological insulators double perovskites: A2TePoO6 (A= Ca, Sr, Ba). J. Appl. Phys. 2017, 122, 224902. [Google Scholar] [CrossRef]
- Hada, M.; Norimatsu, K.; Tanaka, S.; Keskin, S.; Tsuruta, T.; Igarashi, K.; Ishikawa, T.; Kayanuma, Y.; Miller, R.J.D.; Onda, K.; et al. Bandgap modulation in photoexcited topological insulator Bi2Te3 via atomic displacements. J. Chem. Phys. 2016, 145, 024504. [Google Scholar] [CrossRef]
- Annese, E.; Okuda, T.; Schwier, E.F.; Iwasawa, H.; Shimada, K.; Natamane, M.; Taniguchi, M.; Rusinov, I.P.; Kokh, S.V.E.K.A.; Golyashov, V.A.; et al. Electronic and spin structure of the wide-band-gap topological insulator: Nearly stoichiometric Bi2Te2S. Phys. Rev. B 2018, 97, 205113. [Google Scholar] [CrossRef]
- Afsari, M.; Boochani, A.; Hantezadeh, M.; Elahi, S.M. Topological nature in cubic phase of perovskite CsPbI3: By DFT. Solid State Commun. 2017, 259, 10. [Google Scholar] [CrossRef]
- Yalameha, S.; Saeidi, P.; Nourbakhsh, Z.; Vaez, A.; Ramazani, A. Insight into the topological phase and elastic properties of halide perovskites CsSnX3 (X= I, Br, Cl) under hydrostatic pressures. J. Appl. Phys. 2020, 127, 085102. [Google Scholar] [CrossRef]
- Padilha, J.E.; Pontes, R.B.; Schmidt, T.M.; Miwa, R.H.; Fazzio, A. A new class of large band gap quantum spin hall insulators: 2D fluorinated group-IV binary compounds. Sci. Rep. 2016, 6, 26123. [Google Scholar] [CrossRef]
- Wu, L.; Gu, K.; Li, Q. New families of large band gap 2D topological insulators in ethynyl-derivative functionalized compounds. Appl. Surf. Sci. 2019, 484, 1208. [Google Scholar] [CrossRef]
- Gao, C.-L.; Qian, D.; Liu, C.-H.; Jia, J.-F.; Liu, F. Topological edge states and electronic structures of a 2D topological insulator: Single-bilayer Bi (111). Chin. Phys. B 2013, 22, 067304. [Google Scholar] [CrossRef]
- Mahmud, S.; Alam, M.K. Large bandgap quantum spin Hall insulator in methyl decorated plumbene monolayer: A first-principles study. RSC Adv. 2019, 9, 42194. [Google Scholar] [CrossRef]
- Maxwell, C.R. Physical Properties of Polonium. I. Melting Point, Electrical Resistance, Density, and Allotropy. J. Chem. Phys. 1949, 17, 1288. [Google Scholar] [CrossRef]
- Bagnall, K.W.; D’Eye, R.W.M. The preparation of polonium metal and polonium dioxide. J. Chem. Soc. 1954, 4295–4299. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B 1993, 48, 13115. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci 1996, 6, 15. [Google Scholar] [CrossRef]
- Wang, Y.; Perdew, J.P. Correlation hole of the spin-polarized electron gas, with exact small-wave-vector and high-density scaling. Phys. Rev. B 1991, 44, 13298. [Google Scholar] [CrossRef]
- Wang, Y.; Perdew, J.P. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244. [Google Scholar]
- Fu, L.; Kane, C.L. Topological insulators with inversion symmetry. Phys. Rev. B 2007, 76, 045302. [Google Scholar] [CrossRef]
- Jin, H.; Rhim, S.H.; Im, J.; Freeman, A.J. Topological Oxide Insulator in Cubic Perovskite Structure. Sci. Rep. 2013, 3, 1651. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.-J.; Zhao, E.-J.; Xiang, H.-P.; Hao, X.-F.; Liu, X.-J.; Meng, J. Crystal structures and elastic properties of superhard IrN2 and IrN3 from first principles. Phys. Rev. B 2007, 76, 054115. [Google Scholar] [CrossRef]
- Lakes, R. Foam Structures with a Negative Poisson’s Ratio. Science 1987, 235, 1038. [Google Scholar] [CrossRef] [PubMed]
- Evans, K.E. Auxetic polymers: A new range of materials. Endeavour 1991, 15, 170. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).