
PC12 and THP-1 Cell Lines as Neuronal and Microglia Model in Neurobiological Research


Abstract
:1. Introduction
- Screening for toxicity in pharmacological studies of newly synthesized drugs and discovered natural substances prior to in vivo testing;
- Studying the effect of drugs on tissues, cells and their organelles;
- Studying pathways and pathophysiological mechanisms involved in disease onset and progression;
- Production of specific proteins or cellular substances, which are not yet obtained in recombinant form;
- Microbiology and virology (mainly multiplication of intracellular pathogens).
2. Materials and Methods
2.1. Cell Lines
2.2. Coating Culture Plates with Type I Collagen
2.3. Culture Media
2.4. Used Harmful Compounds
2.5. Experimental Design
2.6. Immunofluorescence Staining For Doublecortin
2.7. MTT Assay
2.8. LDH Assay
2.9. DCF-DA Assay
2.10. Fast Halo Assay (FHA)
2.11. Length and Number of Neurites
2.12. Statistical Analysis
3. Results and Discussion
3.1. PC12 Cell Line as Neurobiological Model
3.2. PC12 Cells and Amyloid β (25-35)
3.3. PC12 Cells and Lipopolysaccharide
3.4. Neuronal Features of PC12 Cells after Incubation with Aβ25-35 or LPS
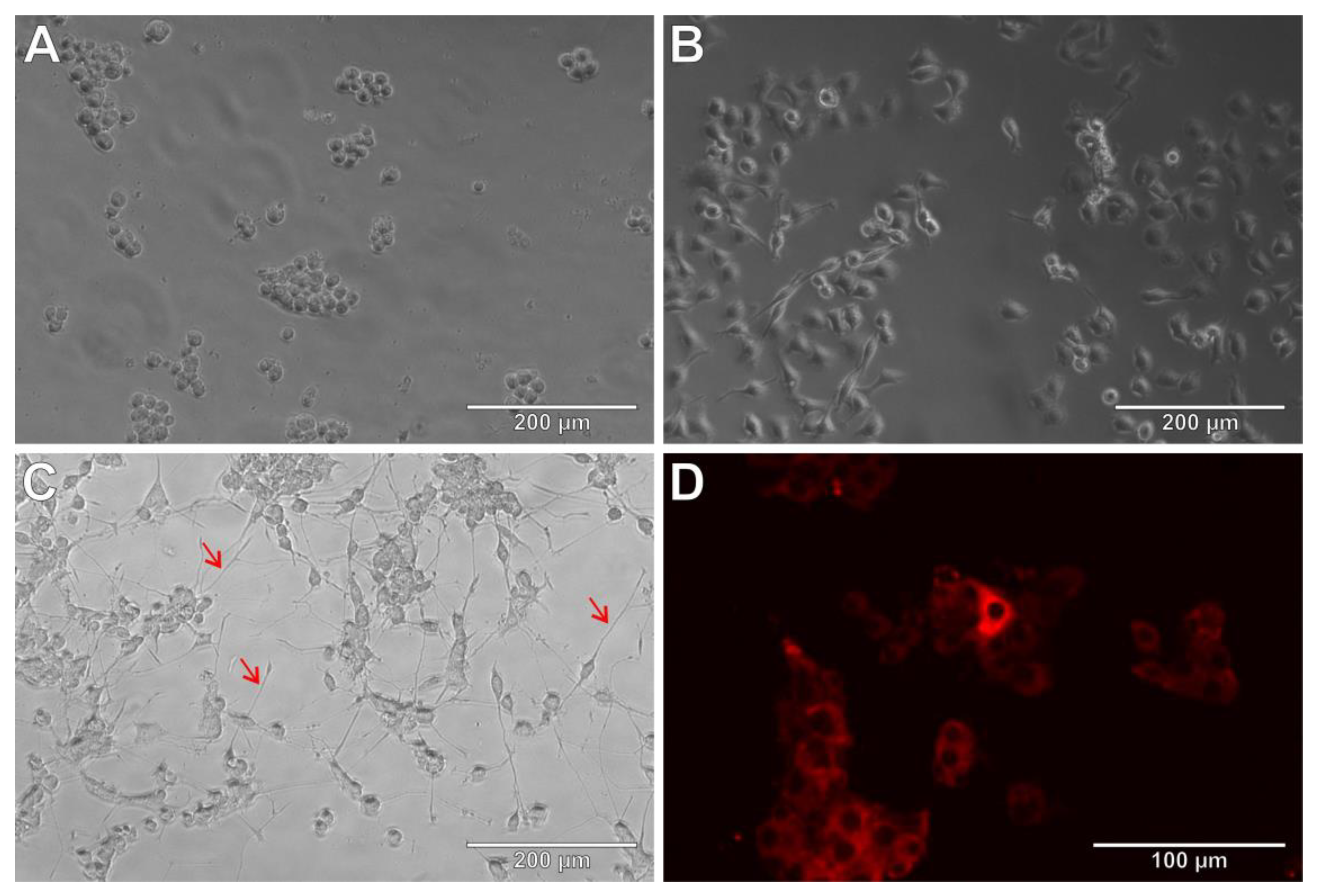
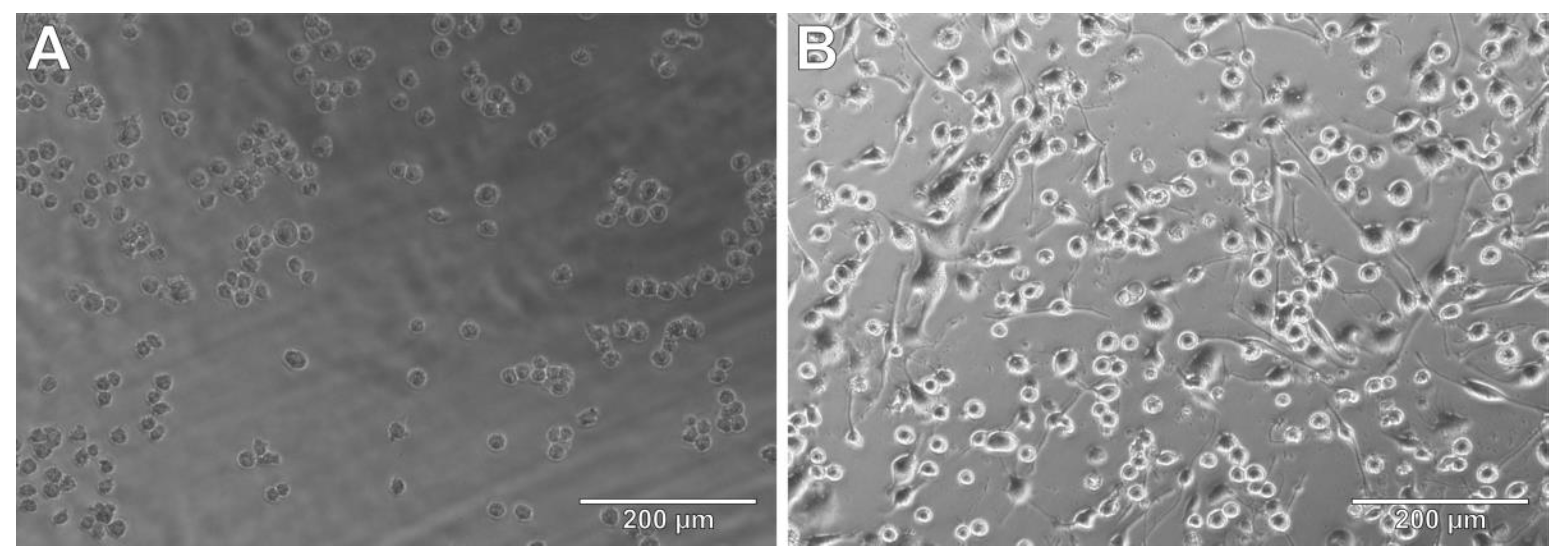
3.5. THP-1 Cells as Microglia Model
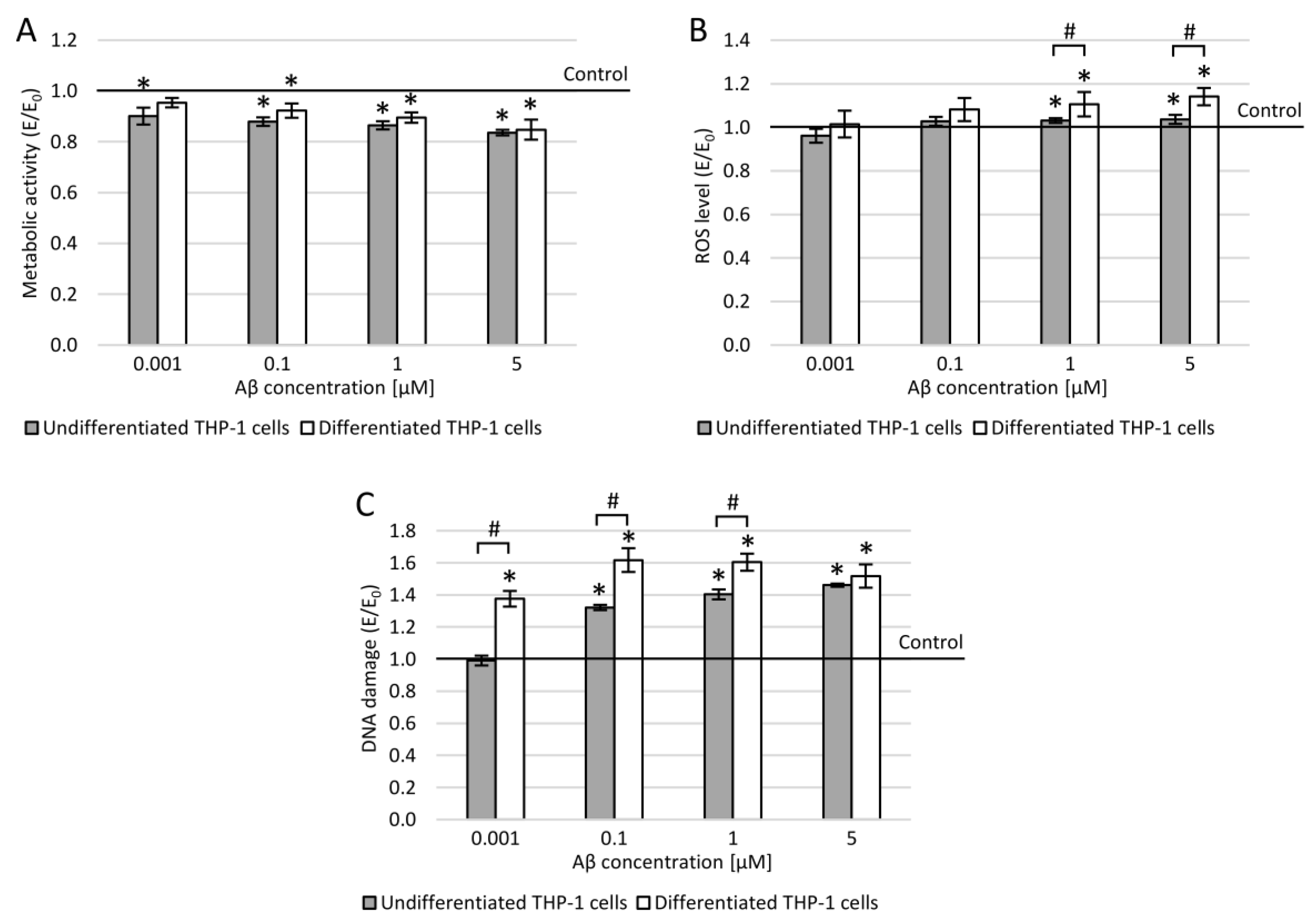
3.6. THP-1 Cells and Amyloid β (25-35)
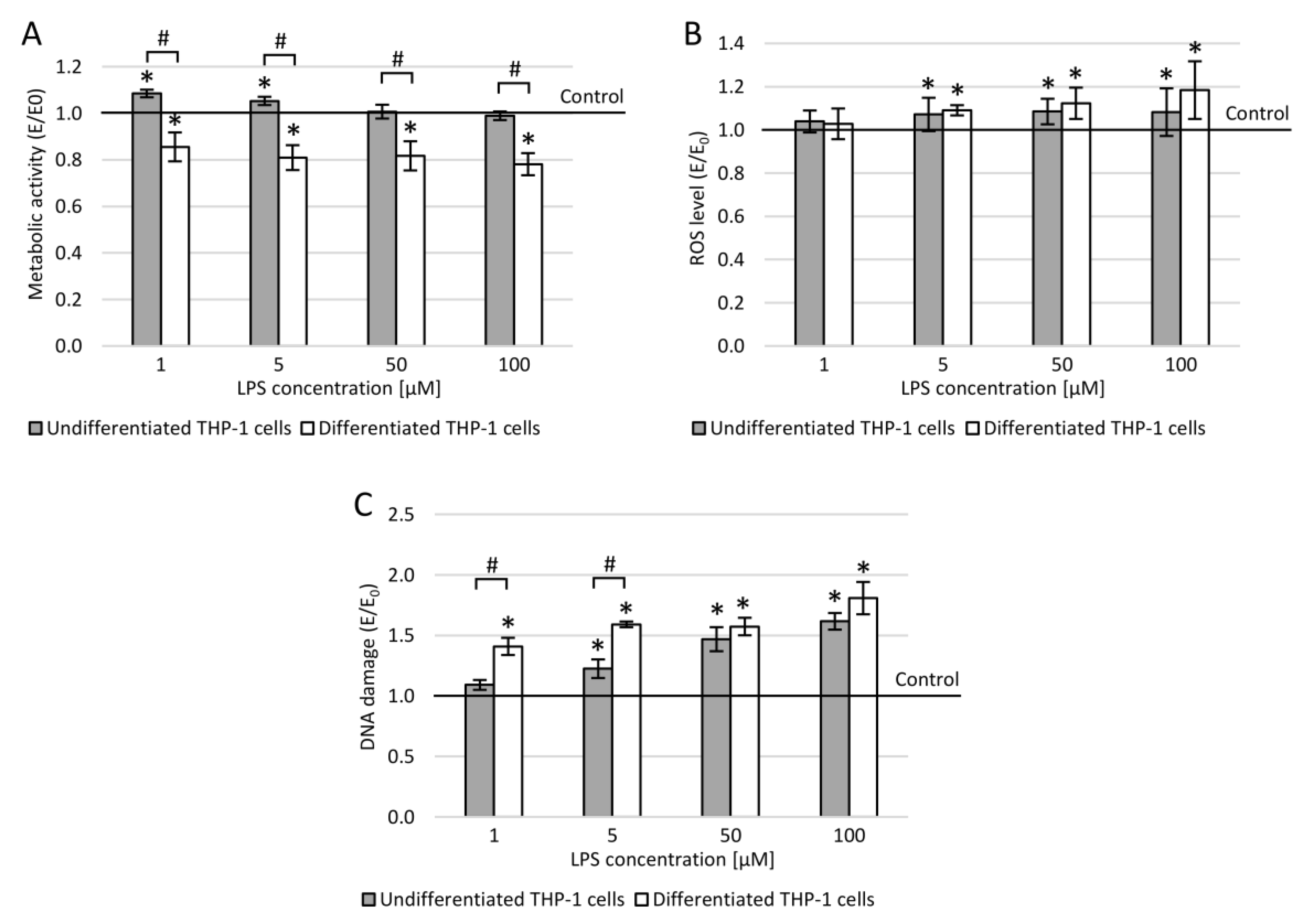
3.7. THP-1 Cells and Lipopolysaccharide
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saeidnia, S.; Manayi, A.; Abdollahi, M. From in vitro Experiments to in vivo and Clinical Studies; Pros and Cons. Curr. Drug Discov. Technol. 2016, 12, 218–224. [Google Scholar] [CrossRef]
- García, Y.; Díaz-Castro, J. Advantages and disadvantages of the animal models v. in vitro studies in iron metabolism: A review. Animal 2013, 7, 1651–1658. [Google Scholar] [CrossRef]
- Chanput, W.; Mes, J.J.; Wichers, H.J. THP-1 cell line: An in vitro cell model for immune modulation approach. Int. Immunopharmacol. 2014, 23, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Spangenberg, E.E.; Green, K.N. Inflammation in Alzheimer’s disease: Lessons learned from microglia-depletion models. Brain. Behav. Immun. 2017, 61, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, M.R.; St-Pierre, M.K.; Wendeln, A.C.; Makoni, N.J.; Gouwens, L.K.; Garrad, E.C.; Sohrabi, M.; Neher, J.J.; Tremblay, M.E.; Combs, C.K. Inflammatory mechanisms in neurodegeneration. J. Neurochem. 2019, 149, 562–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batista, C.R.A.; Gomes, G.F.; Candelario-Jalil, E.; Fiebich, B.L.; de Oliveira, A.C.P. Lipopolysaccharide-Induced Neuroinflammation as a Bridge to Understand Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Zhou, J. wei Neuroinflammation in the central nervous system: Symphony of glial cells. Glia 2019, 67, 1017–1035. [Google Scholar] [CrossRef]
- Welling, M.M.; Nabuurs, R.J.A.; Van Der Weerd, L. Potential role of antimicrobial peptides in the early onset of Alzheimer’s disease. Alzheimer’s Dement. 2015, 11, 51–57. [Google Scholar] [CrossRef]
- Moir, R.D.; Lathe, R.; Tanzi, R.E. The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 1602–1614. [Google Scholar] [CrossRef]
- Dewapriya, P.; Li, Y.X.; Himaya, S.W.A.; Pangestuti, R.; Kim, S.K. Neoechinulin A suppresses amyloid-β oligomer-induced microglia activation and thereby protects PC-12 cells from inflammation-mediated toxicity. Neurotoxicology 2013, 35, 30–40. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Cai, Z.; Hussain, M.D.; Yan, L.J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 307–321. [Google Scholar] [CrossRef]
- Rajendran, L.; Paolicelli, R.C. Microglia-mediated synapse loss in Alzheimer’s disease. J. Neurosci. 2018, 38, 2911–2919. [Google Scholar] [CrossRef] [Green Version]
- Slanzi, A.; Iannoto, G.; Rossi, B.; Zenaro, E.; Constantin, G. In vitro Models of Neurodegenerative Diseases. Front. Cell Dev. Biol. 2020, 8, 328. [Google Scholar] [CrossRef]
- Wiatrak, B.; Kubis-Kubiak, A.; Piwowar, A.; Barg, E. PC12 Cell Line: Cell Types, Coating of Culture Vessels, Differentiation and Other Culture Conditions. Cells 2020, 9, 958. [Google Scholar] [CrossRef]
- Yang, H.H.; Zhang, C.; Lai, S.H.; Zeng, C.C.; Liu, Y.J.; Wang, X.Z. Isoliquiritigenin Induces Cytotoxicity in PC-12 Cells In Vitro. Appl. Biochem. Biotechnol. 2017, 183, 1173–1190. [Google Scholar] [CrossRef]
- Mendonça, L.M.; da Silva Machado, C.; Correia Teixeira, C.C.; Pedro de Freitas, L.A.; Pires Bianchi, M.D.L.; Greggi Antunes, L.M. Curcumin reduces cisplatin-induced neurotoxicity in NGF-differentiated PC12 cells. Neurotoxicology 2013, 34, 205–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, Y.; Sakagami, H.; Shi, H.; Abe, T.; Tamura, N.; Takeshima, H.; Horie, N.; Kaneko, T.; Shiratsuchi, H.; Kaneko, T. Partial protection of paclitaxel-induced neurotoxicity by antioxidants. In Vivo 2018, 32, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Xu, B.; Que, H.; Lin, Q.; Lv, S.; Liu, S. Neurons derived from PC12 cells have the potential to develop synapses with primary neurons from rat cortex. Acta Neurobiol. Exp. 2006, 66, 105–112. [Google Scholar]
- Christen, V.; Rusconi, M.; Crettaz, P.; Fent, K. Developmental neurotoxicity of different pesticides in PC-12 cells in vitro. Toxicol. Appl. Pharmacol. 2017, 325, 25–36. [Google Scholar] [CrossRef]
- Malagelada, C.; Greene, L.A. PC12 Cells as a model for parkinson’s disease research. In Parkinson’s Disease; Nass, R., Przedborski, S., Eds.; Academic Press: Cambridge, MA, USA, 2008; pp. 375–387. [Google Scholar]
- Jesky, R.; Chen, H. The neuritogenic and neuroprotective potential of senegenin against Aβ-induced neurotoxicity in PC 12 cells. BMC Complement. Altern. Med. 2016, 16, 26. [Google Scholar] [CrossRef] [Green Version]
- Eggers, C.; Fujitani, M.; Kato, R.; Smid, S. Novel cannabis flavonoid, cannflavin A displays both a hormetic and neuroprotective profile against amyloid β-mediated neurotoxicity in PC12 cells: Comparison with geranylated flavonoids, mimulone and diplacone. Biochem. Pharmacol. 2019, 169, 113609. [Google Scholar] [CrossRef]
- Bokare, A.M.; Praveenkumar, A.K.; Bhonde, M.; Nayak, Y.; Pal, R.; Goel, R. 5-HT6 Receptor Agonist and Antagonist Against β-Amyloid-Peptide-Induced Neurotoxicity in PC-12 Cells. Neurochem. Res. 2017, 42, 1571–1579. [Google Scholar] [CrossRef]
- Guroff, G. PC12 cells as a model of neuronal differentiation. In Cell Culture in the Neurosciences; Springer: Boston, MA, USA, 1985; pp. 245–272. [Google Scholar]
- Sakagami, H.; Hara, Y.; Shi, H.; Iwama, S.; Nakagawa, M.; Suzuki, H.; Tanaka, K.; Tomoyuki, A.B.E.; Tamura, N.; Takeshima, H.; et al. Change in anticancer drug sensitivity during neuronal differentiation of PC12 cells. In Vivo 2018, 32, 765–770. [Google Scholar] [CrossRef]
- Zhu, D.-J.; Chen, X.-W.; Wang, J.-Z.; Ju, Y.-L.; Ou Yang, M.-Z.; Zhang, W.-J. Proteomic analysis identifies proteins associated with curcumin-enhancing efficacy of irinotecan-induced apoptosis of colorectal cancer LOVO cell. Int. J. Clin. Exp. Pathol. 2014, 7, 1–15. [Google Scholar]
- Zhang, D.; Wang, Z.; Sheng, C.; Peng, W.; Hui, S.; Gong, W.; Chen, S. Icariin Prevents Amyloid Beta-Induced Apoptosis via the PI3K/Akt Pathway in PC-12 Cells. Evidence-Based Complement. Altern. Med. 2015, 235265. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Liu, Y.; Xu, L.; Sha, C.; Zhang, H.; Xu, W. Resveratrol alleviates lipopolysaccharide-induced inflammation in PC-12 cells and in rat model. BMC Biotechnol. 2019, 19, 10. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, G. Matrine protects PC12 cells from lipopolysaccharide-evoked inflammatory injury via upregulation of miR-9. Pharm. Biol. 2020, 58, 314–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Zhang, C.; Zhang, Z.; Dai, Y.; Gu, R.; Jiang, R. Geniposide protects PC12 cells from lipopolysaccharide-evoked inflammatory injury via up-regulation of miR-145-5p. Artif. Cells Nanomed. Biotechnol. 2019, 47, 2875–2881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiatrak, B.; Balon, K. Protective Activity of Aβ on Cell Cultures (PC12 and THP-1 after Differentiation) Preincubated with Lipopolysaccharide (LPS). Mol. Neurobiol. 2020, 58, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Brothers, H.M.; Gosztyla, M.L.; Robinson, S.R. The Physiological Roles of Amyloid-β Peptide Hint at New Ways to Treat Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 118. [Google Scholar] [CrossRef]
- Soscia, S.J.; Kirby, J.E.; Washicosky, K.J.; Tucker, S.M.; Ingelsson, M.; Hyman, B.; Burton, M.A.; Goldstein, L.E.; Duong, S.; Tanzi, R.E.; et al. The Alzheimer’s Disease-Associated Amyloid β-Protein Is an Antimicrobial Peptide. PLoS ONE 2010, 5, e9505. [Google Scholar] [CrossRef]
- Park, E.K.; Jung, H.S.; Yang, H.I.; Yoo, M.C.; Kim, C.; Kim, K.S. Optimized THP-1 differentiation is required for the detection of responses to weak stimuli. Inflamm. Res. 2007, 56, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Lund, M.E.; To, J.; O’Brien, B.A.; Donnelly, S. The choice of phorbol 12-myristate 13-acetate differentiation protocol influences the response of THP-1 macrophages to a pro-inflammatory stimulus. J. Immunol. Methods 2016, 430, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, S.; De Majo, F.; Kim, J.; Trenti, A.; Trevisi, L.; Fadini, G.P.; Bolego, C.; Zandstra, P.W.; Cignarella, A.; Vitiello, L. Convenience versus Biological Significance: Are PMA-Differentiated THP-1 Cells a Reliable Substitute for Blood-Derived Macrophages When Studying in Vitro Polarization? Front. Pharmacol. 2018, 9, 71. [Google Scholar] [CrossRef] [Green Version]
- Daigneault, M.; Preston, J.A.; Marriott, H.M.; Whyte, M.K.B.; Dockrell, D.H. The Identification of Markers of Macrophage Differentiation in PMA-Stimulated THP-1 Cells and Monocyte-Derived Macrophages. PLoS ONE 2010, 5, e8668. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.; Bauler, T.J.; Malik-Kale, P.; Steele-Mortimer, O. The phorbol 12-myristate-13-acetate differentiation protocol is critical to the interaction of THP-1 macrophages with Salmonella Typhimurium. PLoS ONE 2018, 13, e0193601. [Google Scholar] [CrossRef]
- Boutajangout, A.; Wisniewski, T. The innate immune system in Alzheimer’s disease. Int. J. Cell Biol. 2013, 576383. [Google Scholar] [CrossRef] [Green Version]
- Minter, M.R.; Taylor, J.M.; Crack, P.J. The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J. Neurochem. 2016, 136, 457–474. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Advantages | Disadvantages |
|---|---|
|
|
| Undifferentiated PC12 Cells | Differentiated PC12 Cells |
|---|---|
|
|
| Undifferentiated THP-1 Cells | Differentiated THP-1 Cells |
|---|---|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balon, K.; Wiatrak, B. PC12 and THP-1 Cell Lines as Neuronal and Microglia Model in Neurobiological Research. Appl. Sci. 2021, 11, 3729. https://doi.org/10.3390/app11093729
Balon K, Wiatrak B. PC12 and THP-1 Cell Lines as Neuronal and Microglia Model in Neurobiological Research. Applied Sciences. 2021; 11(9):3729. https://doi.org/10.3390/app11093729
Chicago/Turabian StyleBalon, Katarzyna, and Benita Wiatrak. 2021. "PC12 and THP-1 Cell Lines as Neuronal and Microglia Model in Neurobiological Research" Applied Sciences 11, no. 9: 3729. https://doi.org/10.3390/app11093729