
In Silico Studies of Tumor Targeted Peptide-Conjugated Natural Products for Targeting Over-Expressed Receptors in Breast Cancer Cells Using Molecular Docking, Molecular Dynamics and MMGBSA Calculations


Abstract
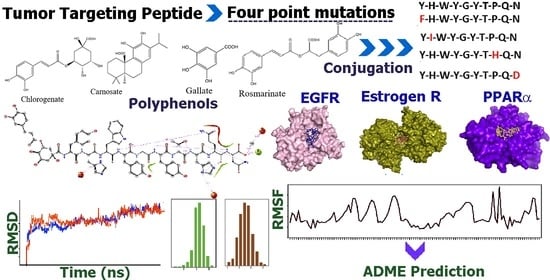
1. Introduction
2. Methods
2.1. Peptide and Conjugate Design
2.2. I-TASSER Studies
2.3. Binding Pocket Analysis
2.4. Receptor-Ligand Docking Studies
2.5. Protein-Ligand Interaction Determination
2.6. Molecular Dynamics Studies
2.7. MMGBSA Energy Calculations
2.8. ADME Studies
3. Results and Discussion
3.1. Anti CP Studies
3.2. I-TASSER Studies
3.3. Receptor Binding Pocket Analysis
3.4. Molecular Docking Studies
3.5. Binding Interaction Analysis with PLIP
3.5.1. Interactions with Estrogen Receptor
3.5.2. Interactions with PPAR-α
3.5.3. Interactions with EGFR
3.6. Molecular Dynamics Studies
3.6.1. Analysis of RMSDs and RMSFs for Ligand-Receptor Complexes
3.6.2. Protein-Ligand Interactions
ER-α
PPAR-α
EGFR
3.7. MM-GBSA Free Energy Studies
4. ADME Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xiao, Y.F.; Jie, M.M.; Li, B.S.; Hu, C.J.; Xie, R.; Tang, B.; Yang, S.M. Peptide-Based Treatment: A Promising Cancer Therapy. J. Immunol. Res. 2015, 2015, e761820. [Google Scholar] [CrossRef] [PubMed]
- Le Joncour, V.; Laakkonen, P. Seek & Destroy, use of targeting peptides for cancer detection and drug delivery. Bioorg. Med. Chem. 2018, 26, 2797–2806. [Google Scholar]
- Scodeller, P.; Asciutto, E.K. Targeting Tumors Using Peptides. Molecules 2020, 25, 808. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Xu, Y. Recent trends and applications of molecular modeling in GPCR-ligand recognition and structure based design. Int. J. Mol. Sci. 2018, 19, 2105. [Google Scholar] [CrossRef]
- Kostrzewa, T.; Sahu, K.K.; Gorska-Ponikowska, M.; Tuszynski, J.A.; Kuban-Jankowska, A. Synthesis of small peptide compounds, molecular docking, and inhibitory activity evaluation against phosphatases PTP1B and SHP2. Drug Des. Devel. Ther. 2018, 12, 4139–4147. [Google Scholar] [CrossRef] [PubMed]
- Feiner, R.C.; Kemker, I.; Krutzke, L.; Allmendinger, E.; Mandell, D.J.; Sewald, N.; Kochanek, S.; Müller, K.M. EGFR-Binding Peptides: From Computational Design towards Tumor-Targeting of Adeno-Associated Virus Capsids. Int. J. Mol. Sci. 2020, 21, 9535. [Google Scholar] [CrossRef] [PubMed]
- Aranda, F.; Vacchelli, E.; Eggermont, A.; Galon, J.; Sautès-Fridman, C.; Tartour, E.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Peptide vaccines in cancer therapy. OncoImmunology 2013, 2, e26621. [Google Scholar] [CrossRef]
- Hoppenz, P.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide-Drug Conjugates and Their Targets in Advanced Cancer Therapies. Front. Chem. 2020, 8, 571. [Google Scholar] [CrossRef]
- Jaracz, S.; Chen, J.; Kuznetsova, L.V.; Ojima, I. Recent Advances in Tumor-Targeting Anticancer Drug Conjugates. Bioorg. Med. Chem. 2005, 13, 5043–5054. [Google Scholar] [CrossRef]
- Cooper, B.M.; Iegre, J.; Donovan, D.H.O.; Halvarsson, M.Ö.; Spring, D.R. Peptides as a platform for targeted therapeutics for cancer: Peptide–drug conjugates (PDCs). Chem. Soc. Rev. 2021, 50, 1480–1494. [Google Scholar] [CrossRef]
- He, R.; Finan, B.; Mayer, J.P.; DiMarchi, R.D. Peptide Conjugates with Small Molecules Designed to Enhance Efficacy and Safety. Molecules 2019, 24, 1855. [Google Scholar] [CrossRef]
- Song, Q.; Chuan, X.; Chen, B.; He, B.; Zhang, H.; Dai, W.; Wang, X.; Zhang, Q. A Smart Tumor Targeting Peptide–Drug Conjugate, PHLIP-SS-DOX: Synthesis and Cellular Uptake on MCF-7 and MCF-7/Adr Cells. Drug Deliv. 2016, 23, 1734–1746. [Google Scholar] [CrossRef]
- Gilad, Y.; Noy, E.; Senderowitz, H.; Albeck, A.; Firer, M.A.; Gellerman, G. Synthesis, biological studies and molecular dynamics of new anticancer RGD-based peptide conjugates for targeted drug delivery. Bioorg. Med. Chem. 2016, 24, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Azqueta, A.; Collins, A. Polyphenols and DNA Damage: A Mixed Blessing. Nutrients 2016, 8, 785. [Google Scholar] [CrossRef]
- Shaikh, A.A.; Braakhuis, A.J.; Bishop, K.S. The Mediterranean Diet and Breast Cancer: A Personalised Approach. Healthcare 2019, 7, 104. [Google Scholar] [CrossRef] [PubMed]
- Bhosale, P.B.; Ha, S.E.; Vetrivel, P.; Kim, H.H.; Kim, S.M.; Kim, G.S. Functions of polyphenols and its anticancer properties in biomedical research: A narrative review. Transl. Cancer Res. 2020, 9, 7619–7631. [Google Scholar] [CrossRef]
- Yesil-Celiktas, O.; Sevimli, C.; Bedir, E.; Vardar-Sukan, F. Inhibitory Effects of Rosemary Extracts, Carnosic Acid and Rosmarinic Acid on the Growth of Various Human Cancer Cell Line. Plant Foods Hum. Nutr. 2010, 65, 158–163. [Google Scholar] [CrossRef]
- Meng, S.; Cao, J.; Feng, Q.; Peng, J.; Hu, Y. Roles of Chlorogenic Acid on Regulating Glucose and Lipids Metabolism: A Review. Evid. Based Complement Alternat. Med. 2013, 2013, 801457. [Google Scholar] [CrossRef]
- Faried, A.; Kurnia, D.; Faried, L.; Usman, N.; Miyazaki, T.; Kato, H.; Kuwano, H. Anticancer effects of gallic acid isolated from Indonesian herbal medicine, Phaleria macrocarpa (Scheff.) Boerl, on human cancer cell lines. Int. J. Oncol. 2007, 30, 605–613. [Google Scholar] [CrossRef]
- Fantini, M.; Benvenuto, M.; Masuelli, L.; Frajese, G.V.; Tresoldi, I.; Modesti, A.; Bei, R. In Vitro and in Vivo Antitumoral Effects of Combinations of Polyphenols, or Polyphenols and Anticancer Drugs: Perspectives on Cancer Treatment. Int. J. Mol. Sci. 2015, 16, 9236–9282. [Google Scholar] [CrossRef]
- Chen, H.; Yao, K.; Nadas, J.; Bode, A.M.; Malakhova, M.; Oi, N.; Li, H.; Lubet, R.A.; Dong, Z. Prediction of Molecular Targets of Cancer Preventing Flavonoid Compounds Using Computational Methods. PLoS ONE 2012, 7, e38261. [Google Scholar] [CrossRef]
- Chen, H.; Yao, K.; Chang, X.; Shim, J.; Kim, H.; Malakhova, M.; Kim, D.; Bode, A.M.; Dong, Z. Computational and Biochemical Discovery of RSK2 as a Novel Target for Epigallocatechin Gallate (EGCG). PLoS ONE 2015, 10, e0130049. [Google Scholar] [CrossRef] [PubMed]
- Aminpour, M.; Montemagno, C.; Tuszynski, J.A. An Overview of Molecular Modeling for Drug Discovery with Specific Illustrative Examples of Applications. Molecules 2019, 24, 1693. [Google Scholar] [CrossRef] [PubMed]
- Salmaso, V.; Moro, S. Bridging Molecular Docking to Molecular Dynamics in Exploring Ligand-Protein Recognition Process: An Overview. Front. Pharmacol. 2018, 9, 923. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Chakraborty, A.; Biswas, A.; Chowdhuri, S. Identification of polyphenols from Broussonetia papyrifera as SARS CoV-2 main protease inhibitors using in silico docking and molecular dynamics simulation approaches. J. Biomol. Struct. Dynamics. 2020, 39, 6747–6760. [Google Scholar] [CrossRef] [PubMed]
- Manivannan, A.; Soundararajan, P.; Park, Y.G.; Sakkiah, S.; Jeong, B.R. Binding Mode Investigation of Polyphenols from Scrophularia Targeting Human Aldose Reductase Using Molecular Docking and Molecular Dynamics Simulations. J. Chem. 2015, 2015, e434256. [Google Scholar] [CrossRef]
- Tan, Y.; Wang, M.; Yang, K.; Chi, T.; Liao, Z.; Wei, P. PPAR-α Modulators as Current and Potential Cancer Treatments. Front. Oncol. 2021, 11, e599995. [Google Scholar] [CrossRef]
- Costa, R.; Shah, A.N.; Santa-Maria, C.A.; Cruz, M.R.; Mahalingam, D.; Carneiro, B.A.; Chae, Y.K.; Cristofanilli, M.; Gradishar, W.J.; Giles, F.J. Targeting epidermal growth factor receptor in triple negative breast cancer: New discoveries and practical insights for drug development. Cancer Treat. Rev. 2017, 53, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Hossein-Nejad-Ariani, H.; Althagafi, E.; Kaur, K. Small Peptide Ligands for Targeting EGFR in Triple Negative Breast Cancer Cells. Sci. Rep. 2019, 9, 2723. [Google Scholar] [CrossRef]
- Skandalis, S.S.; Afratis, N.; Smirlaki, G.; Nikitovic, D.; Theocharis, A.D.; Tzanakakis, G.N.; Karamanos, N.K. Cross-talk between estradiol receptor and EGFR/IGF-IR signaling pathways in estrogen-responsive breast cancers: Focus on the role and impact of proteoglycans. Matrix Biol. 2014, 35, 182–193. [Google Scholar] [CrossRef]
- Jeong, J.; Kim, H.; Choi, J. In Silico Molecular Docking and In Vivo Validation with Caenorhabditis elegans to Discover Molecular Initiating Events in Adverse Outcome Pathway Framework: Case Study on Endocrine-Disrupting Chemicals with Estrogen and Androgen Receptors. Int. J. Mol. Sci. 2019, 20, 1209. [Google Scholar] [CrossRef]
- Vijaykumar, S.; Ptv, L. ACPP: A webserver for prediction and design of anticancer peptides. Int. J. Pept. Res. Ther. 2015, 21, 99–106. [Google Scholar] [CrossRef]
- Tyagi, A.; Kapoor, P.; Kumar, R.; Chaudhary, K.; Gautam, A. Raghava GPS. In Silico Models for Designing and Discovering Novel Anticancer Peptides. Sci. Rep. 2013, 3, 2984. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System, Version 2.0; Schrödinger LLC: New York, NY, USA, 2020.
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Roy, A.; Zhang, Y. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013, 29, 2588–2595. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhou, Y.; Tanaka, I.; Yao, M. Roll: A new algorithm for the detection of protein pockets and cavities with a rolling probe sphere. Bioinformatics 2010, 26, 46–52. [Google Scholar] [CrossRef]
- Cipolletti, M.; Fernandez, V.S.; Montalesi, E.; Marino, M.; Fiocchetti, M. Beyond the Antioxidant Activity of Dietary Polyphenols in Cancer: The Modulation of Estrogen Receptors (ERs) Signaling. Int. J. Mol. Sci. 2018, 19, 2624. [Google Scholar] [CrossRef] [PubMed]
- Sierra, M.L.; Beneton, V.; Boullay, A.B.; Boyer, T.; Brewster, A.G.; Donche, F.; Forest, M.C.; Fouchet, M.H.; Gellibert, F.J.; Grillot, D.A.; et al. Substituted 2-[(4-Aminomethyl)Phenoxy]-2-Methylpropionic Acid PPARα Agonists. 1. Discovery of a Novel Series of Potent HDLc Raising Agent. J. Med. Chem. 2007, 50, 685–695. [Google Scholar] [CrossRef]
- Xu, G.; Abad, M.C.; Connolly, P.J.; Neeper, M.P.; Struble, G.T.; Springer, B.A.; Emanuel, S.L.; Pandey, N.; Gruninger, R.H.; Adams, M.; et al. 4-Amino-6-arylamino-pyrimidine-5-carbaldehyde hydrazones as potent ErbB-2/EGFR dual kinase inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 4615–4619. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Schneidman, D.; Nussinov, R.; Wolfson, H.J. Efficient Unbound Docking of Rigid Molecules. In Proceedings of the Algorithms in Bioinformatics, Second International Workshop, WABI, Rome, Italy, 17–21 September 2002; Volume 2542, pp. 185–200. [Google Scholar]
- Mashiach, E.; Schneidman-Duhovny, D.; Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: A Web Server for Fast Interaction Refinement in Molecular Docking. Nucleic Acids Res. 2008, 36, W229–W232. [Google Scholar] [CrossRef] [PubMed]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef]
- Pérez-Benito, L.; Keränen, H.; van Vlijmen, H.; Tresadern, G. Predicting Binding Free Energies of PDE2 Inhibitors. The Difficulties of Protein Conformation. Sci. Rep. 2018, 8, 4883. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, P.P.; Das, B.K.; Soorya, E.; Chakraborty, D. Effect of hydrophobic and hydrogen bonding interactions on the potency of ss-alanine analogs of G-protein coupled glucagon receptor inhibitors. Proteins 2020, 88, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 440–461. [Google Scholar] [CrossRef]
- Du, J.; Sun, H.; Xi, L.; Li, J.; Yang, Y.; Liu, H.; Yao, X. Molecular modeling study of checkpoint kinase 1 inhibitors by multiple docking strategies and prime/MM GBSA calculation. J. Comp. Chem. 2011, 32, 2800–2809. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Chen, X.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- Puranik, N.V.; Srivastava, P.; Bhatt, G.; Mary, D.J.S.J.; Limaye, A.M.; Sivaraman, J. Determination and Analysis of Agonist and Antagonist Potential of Naturally Occurring Flavonoids for Estrogen Receptor (ERα) by Various Parameters and Molecular Modelling Approach. Sci. Rep. 2019, 9, 7450. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, S.; Xu, W.; Wang, R.; Chou, K. Design novel dual agonists for treating type-2 diabetes by targeting peroxisome proliferator-activated receptors with core hopping approach. PLoS ONE 2012, 7, e38546. [Google Scholar] [CrossRef]
- Chen, G.; Seukep, A.J.; Guo, M. Recent advances in molecular docking for the research and discovery of potential marine drugs. Mar. Drugs. 2020, 18, 545. [Google Scholar] [CrossRef]
- Grahl, M.V.C.; Alcará, A.M.; Perin, A.P.A.; Moro, C.F.; Pinto, É.S.M.; Feltes, B.C.; Ghilardi, I.M.; Rodrigues, F.V.F.; Dorn, M.; da Costa, J.C.; et al. Evaluation of drug repositioning by molecular docking of pharmaceutical resources available in the brazilian healthcare systems against SARS-CoV2. Inform. Med. Unlocked 2021, 23, 100539. [Google Scholar] [CrossRef]
- Bianco, G.; Forli, S.; Goodsell, D.S.; Olson, A.J. Covalent docking using autodock: Two-point attractor and flexible side chain methods. Protein Sci. 2016, 25, 295–301. [Google Scholar] [CrossRef]
- Yudt, M.R.; Vorojeikina, D.; Zhong, L.; Skafar, D.F.; Sasson, S.; Gasiewicz, T.A.; Notides, A.C. Function of estrogen receptor tyrosine 537 in hormone binding, DNA binding and transactivation. Biochemistry 1999, 38, 14146–14156. [Google Scholar] [CrossRef] [PubMed]
- Grande, F.; Rizzuti, B.; Occhiuzzi, M.A.; Ioele, G.; Casacchia, T.; Gelmini, F.; Guzzi, R.; Garofalo, A.; Statti, G. Identification by Molecular Docking of Homoisoflavones from Leopoldia comosa as Ligands of Estrogen Receptors. Molecules 2018, 23, 894. [Google Scholar] [CrossRef]
- Lambrinidis, G.; Halabalaki, M.; Katsanou, E.S.; Skaltsounis, A.-L.; Alexis, M.N.; Mikros, E. The estrogen receptor and polyphenos: Molecular simulation studies of their interactions, a review. Environ. Chem. Lett. 2006, 4, 159–174. [Google Scholar] [CrossRef]
- Youssef, J.; Badr, M. Peroxisome proliferator-activated receptors and cancer: Challenges and opportunities. Br. J. Pharmacol. 2011, 164, 68–82. [Google Scholar] [CrossRef]
- Lalloyer, F.; Staels, B. Fibrates, glitazones, and peroxisome proliferator-activated receptors. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Varga, T.; Czimmerer, Z.; Nagy, L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim. Biophys. Acta 2011, 1812, 1007–1022. [Google Scholar] [CrossRef]
- Cronet, P.; Petersen, J.F.; Folmer, R.; Blomberg, N.; Sjöblom, K.; Karlsson, U.; Lindstedt, E.L.; Bamberg, K. Structure of the PPARα and –γ ligand binding domain in complex with AZ 242; ligand selectivity and agonist activation in the PPAR family. Structure 2001, 9, 699–706. [Google Scholar] [CrossRef]
- Ugwu, D.I.; Okoro, U.C.; Mishra, N.K.; Okafor, S.N. Novel Phenoxazinones as Potent Agonist of PPAR-α: Design, Synthesis, Molecular Docking and in Vivo Studies. Lipids Health Dis. 2018, 17, 120. [Google Scholar] [CrossRef]
- Jura, N.; Zhang, X.; Endres, N.F.; Seeliger, M.A.; Schindler, T.; Kuriyan, J. Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol. Cell 2011, 42, 9–22. [Google Scholar] [CrossRef]
- Kumar, R.; Zakharov, M.N.; Khan, S.H.; Miki, R.; Jang, H.; Toraldo, G.; Singh, R.; Bhasin, S.; Jasuja, R. The Dynamic Structure of the Estrogen Receptor. J. Amino Acids 2011, 2011, 812540. [Google Scholar] [CrossRef]
- Brzozowski, A.M.; Pike, A.C.W.; Dauter, Z.; Hubbard, R.E.; Bonn, T.; Engström, O.; Öhman, L.; Greene, G.L.; Gustafsson, J.; Carlquist, M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997, 389, 753–758. [Google Scholar] [CrossRef]
- Farooq, A. Structural and functional diversity of estrogen receptor ligands. Curr. Top. Med. Chem. 2015, 15, 1372–1384. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xu, H.E.; Lambert, M.H.; Montana, V.G.; Plunket, K.D.; Moore, L.B.; Collins, J.L.; Oplinger, J.A.; Kliewer, S.A.; Gampe, R.T.; McKee, D.D.; et al. Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 13919–13924. [Google Scholar] [CrossRef]
- Martin-Fernandez, M.L.; Clarke, D.T.; Roberts, S.K.; Zanetti-Domingues, L.C.; Gervasio, F.L. Structure and Dynamics of the EGF Receptor as Revealed by Experiments and Simulations and its Relevance to Non-Small Cell Lung Cancer. Cells 2019, 8, 316. [Google Scholar] [CrossRef] [PubMed]
- Hasenahuer, M.A.; Barletta, G.P.; Fernandez-Alberti, S.; Parisi, G.; Fornasari, M.S. Pockets as structural descriptors of EGFR kinase conformations. PLoS ONE 2017, 12, e0189147. [Google Scholar] [CrossRef] [PubMed]
- Durai, P.; Govindaraj, R.G.; Choi, S. Structure and dynamic behavior of Toll-like receptor 2 subfamily triggered by malarial glycosylphosphatidylinostiols of Plasmodium falciparum. FEBS J. 2013, 280, 6196–6212. [Google Scholar] [CrossRef]
- Kambia, N.; Farce, A.; Belarbi, K.; Gressier, B.; Luyckx, M.; Chavatte, P.; Dine, T. Dokcking Study: PPARs interaction with selected alternative plasticizers to di(2-ethylhexyl) phthalate. J. Enzyme Inhib. Med. Chem. 2016, 13, 448–455. [Google Scholar]
- Ahinko, M.; Niinivehmas, S.; Jokinen, E.; Pentikäinen, O.T. Suitability of MMGBSA for the selection of correct ligand binding modes from docking results. Chem. Biol. Drug Des. 2019, 93, 522–538. [Google Scholar] [CrossRef]
- Lokhande, K.B.; Ballav, S.; Yadav, R.S.; Venkateswara Swamy, K.; Basu, S. Probing intermolecular interactions and binding stability of kaempferol, quercetin and resveratrol derivatives with PPAR-γ: Docking, molecular dynamics and MM/GBSA approach to reveal potent PPAR- γ agonist against cancer. J. Biomol. Struc. Dyn. 2020, 1–11. [Google Scholar] [CrossRef]
- Sharma, V.K.; Nandekar, P.P.; Sangamwar, A.; Pérez-Sánchez, H.; Agarwal, S.M. Structure guided design and binding analysis of EGFR inhibiting analogues of erlotinib and AEE788 using ensemble docking, molecular dynamics and MMGBSA. RSC Adv. 2016, 6, 65725–65735. [Google Scholar] [CrossRef]
- Al-Anazi, M.; Al-Najjar, B.O.; Khairuddean, M. Structure-based drug design studies toward the discovery of novel chalcone derivatives as potential epidermal growth factor receptor (EGFR) inhibitors. Molecules 2018, 23, 3203. [Google Scholar] [CrossRef] [PubMed]
- Morcoss, M.M.; Abdelhafez, E.S.M.N.; Ibrahem, R.A.; Abdel-Rahman, H.M.; Abdel-Aziz, M.; Abou El-Ella, D.A. Design, synthesis, mechanistic studies and in silico ADME predictions of benzimidazole derivatives as novel antifungal agents. Bioorg. Chem. 2020, 101, 103956. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. ADME-SAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.D.; Kumar, S.P.; Pandya, H.A.; Solanki, H.A. MDCKpred: A web-tool to calculate MDCK permeability coefficient of small molecule using membrane-interaction chemical features. Toxicol. Mech. Methods 2018, 28, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Chambers, P.; Wipf, P.; Xie, X.Q.; Englert, D.; Weber, S. Lipophilicity screening of novel drug-like compounds and comparison of cLogP. J. Chromatogr. A 2012, 1258, 161–167. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Original Peptide (Pep) | YHWYGYTPQN |
| Mutation 1 (Mut1) | FHWYGYTPQN |
| Mutation 2 (Mut2) | YIWYGYTPQN |
| Mutation 3 (Mut3) | YHWYGYTHQN |
| Mutation 4 (Mut4) | YHWYGYTPQD |
| A | B | C | D |
|---|---|---|---|
 |  |  |  |
 |  |  |  |
 |  |  |  |
 |  |  |  |
 |  |  |  |
| Peptide Sequence | Mutation Position | SVM Score | Hydrophobicity | Hydropathicity | Hydrophilicity | pI |
| YHWYGYTPQN | None | 0.88 | −0.14 | −1.77 | −1.08 | 7.08 |
| FHWYGYTPQN | 1 | 1.00 | −0.08 | −1.36 | −1.10 | 7.09 |
| YIWYGYTPQN | 2 | 0.77 | −0.03 | −1.00 | −1.21 | 5.87 |
| YHWYGYTHQN | 8 | 0.82 | −0.17 | −1.93 | −1.13 | 7.25 |
| YHWYGYTPQD | 10 | 0.85 | −0.15 | −1.77 | −0.80 | 5.09 |
| Peptide Sequence | Secondary Structure | Solvent Accessibility/Residue in the Sequence * | C Score | |||
| YHWYGYTPQN | CCCCCCCCCC | 7-4-3-4-4-3-4-4-7-8 | −0.58 | |||
| FHWYGYTPQN | CCCCCCCCCC | 6-4-3-4-4-3-4-4-7-8 | −0.56 | |||
| YIWYGYTPQN | CCSSCCCCCC | 6-3-2-4-4-3-4-4-7-8 | −0.54 | |||
| YHWYGYTHQN | CCCCCCCCCC | 7-4-3-4-4-3-4-4-7-8 | −0.68 | |||
| YHWYGYTPQD | CCCCCCCCCC | 7-4-3-4-4-3-4-4-7-8 | −0.46 | |||
| ER-α | |||||
| Peptide Sequence Conjugated | Carnosic Acid | Chlorogenic Acid | Gallic Acid | Rosmarinic Acid | Neat Peptide (Unconjugated) |
| YHWYGYTPQN (pep) | −8.7 | −9.8 | −8.8 | −10.3 | −10.0 |
| FHWYGYTPQN (Mut1) | −7.8 | −10.2 | −9.0 | −8.7 | −8.6 |
| YIWYGYTPQN (Mut2) | −7.8 | −9.6 | −9.5 | −8.6 | −8.6 |
| YHWYGYTHQN (Mut3) | −10.0 | −9.1 | −8.1 | −8.2 | −10.0 |
| YHWYGYTPQD (Mut4) | −9.0 | −10.3 | −10.2 | −9.4 | −9.1 |
| PPAR-α | |||||
| Peptide Sequence Conjugated | Carnosic Acid | Chlorogenic Acid | Gallic Acid | Rosmarinic Acid | Neat Peptide (Unconjugated) |
| YHWYGYTPQN (pep) | −7.1 | −8.1 | −8.3 | −9.2 | −8.9 |
| FHWYGYTPQN (Mut1) | −6.5 | −7.0 | −8.3 | −9.5 | −9.1 |
| YIWYGYTPQN (Mut2) | −6.2 | −7.1 | −9.2 | −5.4 | −7.6 |
| YHWYGYTHQN (Mut3) | −6.5 | −7.2 | −7.4 | −7.6 | −8.9 |
| YHWYGYTPQD (Mut4) | −7.0 | −6.8 | −8.6 | −8.3 | −9.1 |
| EGFR | |||||
| Peptide Sequence Conjugated | Carnosic Acid | Chlorogenic Acid | Gallic Acid | Rosmarinic Acid | Neat Peptide (Unconjugated) |
| YHWYGYTPQN (pep) | −8 | −8.5 | −8.3 | −8.4 | −9.5 |
| FHWYGYTPQN (Mut1) | −8.3 | −8.3 | −7.7 | −9.1 | −7.7 |
| YIWYGYTPQN (Mut2) | −7.2 | −8.2 | −7.7 | −8.4 | −8.7 |
| YHWYGYTHQN (Mut3) | −8.6 | −7.4 | −7.4 | −7.4 | −8.0 |
| YHWYGYTPQD (Mut4) | −8.1 | −7.5 | −8.4 | −8.8 | −8.3 |
| Estrogen-Alpha Receptor | |||||
|---|---|---|---|---|---|
| Compound | Avg. ΔG Binding Energy (kcal/mol) | Avg Electrostatic Energy (kcal/mol) | Avg. H-Bond Energy (kcal/mol) | Avg. Lipophilic Energy (kcal/mol) | Avg. vdW Energy (kcal/mol) |
| Pep | −61.90 | −37.11 | −6.40 | −15.94 | −71.33 |
| Mut1 | −37.97 | −24.44 | −2.52 | −11.21 | −36.98 |
| Mut2 | −70.94 | −37.61 | −5.45 | −17.87 | −69.43 |
| Mut3 | −70.62 | −39.03 | −5.48 | −18.91 | −61.08 |
| Mut4 | −108.90 | −54.87 | −6.72 | −32.92 | −98.60 |
| RMAPep | −59.98 | −38.10 | −6.70 | −15.77 | −70.52 |
| GLAMut1 | −45.97 | −23.47 | −2.11 | −8.74 | −45.23 |
| RMAMut1 | −41.62 | −30.82 | −3.29 | −12.04 | −42.22 |
| GLAMut2 | −47.28 | −30.05 | −2.89 | −11.53 | −42.30 |
| CGAMut4 | −80.92 | −61.98 | −6.34 | −18.01 | −72.00 |
| CSAMut4 | −78.35 | −32.18 | −3.71 | −22.93 | −80.60 |
| PPAR-Alpha Receptor | |||||
| Pep | −69.14 | −18.34 | −4.73 | −20.88 | −64.80 |
| Mut1 | −67.02 | −16.01 | −3.72 | −21.31 | −66.31 |
| Mut2 | −67.78 | −5.03 | −3.37 | −20.63 | −64.29 |
| Mut3 | −63.39 | −17.82 | −4.49 | −18.88 | −61.90 |
| Mut4 | −63.02 | −16.45 | −3.66 | −19.56 | −62.26 |
| RMAPep | −67.44 | −20.33 | −4.29 | −20.60 | −64.76 |
| GLAMut1 | −71.47 | −33.03 | −4.99 | −21.30 | −66.18 |
| RMAMut1 | −65.35 | −28.73 | −3.32 | −21.03 | −65.46 |
| GLAMut2 | −67.35 | −43.97 | −4.39 | −22.28 | −69.34 |
| CGAMut4 | −68.73 | −16.27 | −4.52 | −20.91 | −66.46 |
| CSAMut4 | −40.20 | −9.80 | −1.95 | −12.81 | −39.70 |
| EGFR Receptor | |||||
| Pep | −174.54 | −48.74 | −8.32 | −30.00 | −114.43 |
| Mut1 | −194.22 | −98.96 | −10.01 | −33.74 | −128.01 |
| Mut2 | −173.50 | −54.84 | −8.41 | −29.92 | −113.84 |
| Mut3 | −173.36 | −40.40 | −8.18 | −32.39 | −116.54 |
| Mut4 | −188.66 | −69.22 | −8.52 | −33.58 | −124.70 |
| RMAPep | −63.04 | −37.18 | −3.73 | −16.14 | −53.81 |
| GLAMut1 | −175.16 | −59.21 | −8.33 | −30.74 | −118.13 |
| RMAMut1 | −49.77 | −9.41 | −4.12 | −12.13 | −60.20 |
| GLAMut2 | −173.75 | −39.93 | −7.70 | −32.65 | −117.88 |
| CGAMut4 | −68.30 | −49.13 | −4.76 | −13.82 | 429.28 |
| CSAMut4 | −46.60 | −23.03 | −1.87 | −15.77 | −44.96 |
| Compound | Log P at pH 7.4 | MDCK Permeability (cm/s) | hERG Blocker | Pgp Inhibitor/ Substrate | CYP1A2 Substrate /or Inhibitor |
|---|---|---|---|---|---|
| Pep (Y-H-W-Y-G-Y-T-P-Q-N) | −1.698 | 2.5 × 10−6 | No | No/No | No/No |
| Mut 1 (F-H-W-Y-G-Y-T-P-Q-N) | −0.962 | 2.9 × 10−6 | No | No/Yes | No/No |
| Mut 2 (Y-I-W-Y-G-Y-T-P-Q-N) | −0.407 | 3.2 × 10−6 | No | No/Yes | No/No |
| Mut 3 (Y-H-W-Y-G-Y-T-H-Q-N) | −2.112 | 2 × 10−6 | No | No/Yes | No/No |
| Mut 4 (Y-H-W-Y-G-Y-T-P-Q-D) | −1.487 | 1.9 × 10−6 | No | No/Yes | No/No |
| Rosmarinate-Peptide | 0.046 | 1.9 × 10−6 | No | No/Yes | No/No |
| Gallate-Mut 1 | −0.565 | 2.2 × 10−6 | No | No/Yes | No/No |
| Rosmarinate-Mutation 1 | 0.452 | 2.1 × 10−6 | No | No/Yes | No/No |
| Gallate-Mut 2 | −0.070 | 2.1 × 10−6 | No | No/Yes | No/No |
| Chlorogenate-Mut 4 | −0.089 | 1.4 × 10−6 | No | No/Yes | No/No |
| Carnosate-Mut 4 | 2.145 | 1.8 × 10−6 | No | No/Yes | No/No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hart, L.R.; Lebedenko, C.G.; Mitchell, S.M.; Daso, R.E.; Banerjee, I.A. In Silico Studies of Tumor Targeted Peptide-Conjugated Natural Products for Targeting Over-Expressed Receptors in Breast Cancer Cells Using Molecular Docking, Molecular Dynamics and MMGBSA Calculations. Appl. Sci. 2022, 12, 515. https://doi.org/10.3390/app12010515
Hart LR, Lebedenko CG, Mitchell SM, Daso RE, Banerjee IA. In Silico Studies of Tumor Targeted Peptide-Conjugated Natural Products for Targeting Over-Expressed Receptors in Breast Cancer Cells Using Molecular Docking, Molecular Dynamics and MMGBSA Calculations. Applied Sciences. 2022; 12(1):515. https://doi.org/10.3390/app12010515
Chicago/Turabian StyleHart, Lucy R., Charlotta G. Lebedenko, Saige M. Mitchell, Rachel E. Daso, and Ipsita A. Banerjee. 2022. "In Silico Studies of Tumor Targeted Peptide-Conjugated Natural Products for Targeting Over-Expressed Receptors in Breast Cancer Cells Using Molecular Docking, Molecular Dynamics and MMGBSA Calculations" Applied Sciences 12, no. 1: 515. https://doi.org/10.3390/app12010515
APA StyleHart, L. R., Lebedenko, C. G., Mitchell, S. M., Daso, R. E., & Banerjee, I. A. (2022). In Silico Studies of Tumor Targeted Peptide-Conjugated Natural Products for Targeting Over-Expressed Receptors in Breast Cancer Cells Using Molecular Docking, Molecular Dynamics and MMGBSA Calculations. Applied Sciences, 12(1), 515. https://doi.org/10.3390/app12010515