3.1. The Theoretical Model
We now consider some effects of a solid matrix of inert atoms or molecules at cryogenic temperatures on embedded dilute alkali atoms. In this section we estimate the potential energy of a guest alkali atom in the solid matrix, starting from tabulated values of the relevant parameters, obtained experimentally or theoretically calculated. Our aim here is to obtain estimates of the order of magnitude of the potential, rather than precise values for specific cases, using simple expressions of the dependence of the potentials from the distance, depending from a small number of parameters, and that can be easily adapted to different atom–matrix combinations. We base our analysis on three different analytical models for the potential energy curve of the doping alkali atom and one atom or molecule of the matrix; specifically, we will consider Argon, Neon, and parahydrogen as matrices. The models considered and compared are the Morse potential [
36,
37], the Buckingham–Hill potential [
36,
38,
39], and the model used by Buck et al for H
2-H
2 or D
2-H
2 interactions [
40], and then used also for the Li-H
2 potential [
41,
42]). Our analysis will refer to only a specific peak of the triplet structure of the spectrum, in particular the non-shifted one as described in the previous section.
Evaluating these potentials through simple analytical expressions is an important first step for estimating the contribution to the inhomogeneous linewidth of the energy levels of the dilute guest atoms in the matrix due to their interaction energy with the matrix. In fact, the actual potential energy depends on the position of the atoms in the solid crystal, which can be (slightly) different from atom to atom, yielding different energy-level shifts; this eventually yields a relevant contribution to the resulting inhomogeneous width of the spectral lines.
In the case of our interest, the Buckingham–Hill and the Morse potentials can give a reliable prediction only in different distance ranges: large distances for the Buckingham–Hill potential, and short distances for the Morse potential. More sophisticated models, such as the one proposed by Buck et al., have been used in the literature for the Li-H
2 potential as well as for the H
2-H
2 potential [
40,
41], but they involve a higher number of parameters; we will consider this model for the Li-H
2 potential at the end of this subsection.
As we will now discuss in detail, both of them can be in general necessary in order to evaluate the potential felt by the alkali atom in the solid matrix using a small number of known and tabulated values of the relevant parameters for the potential. It can be also helpful to use an appropriate combination of both models in order to obtain an approximated analytical expression of the actual potential, assuming that the alkali atom substitutes one of the atoms or molecules of the solid matrix. This potential, together with our experimental spectroscopical data of
Section 2, will be then exploited in
Section 3.2 to discuss the consequent contribution to the inhomogeneous broadening of the alkali-atom spectral lines (in particular, of the ground state, involved as initial state of the proposed transition scheme for the axion detection discussed in the Introduction.
We first consider the case of the doped crystal experimentally analyzed in the previous section, that is dilute Rubidium atoms in a Argon or Neon solid matrix, comparing the theoretical results with our experimental data reported in
Section 2. Secondly, we will consider the case of dilute Lithium atoms in a parahydrogen (p-H
2) solid matrix, obtaining insights that this alkali–matrix system yields quite smaller interaction energies, at least of the ground state. We finally suggest that this could be a good guest-atoms–matrix combination for the proposed axion detection scheme.
Both the inert-gas atoms of the cold matrix and the doping alkali atoms have a spherical electronic structure; thus, outside the wavefunction overlap region, the only potential is the van der Waals potential, with a leading term scaling with the distance as
(dipolar two-body dispersion interaction energy) [
43,
44,
45]. We can thus write the total two-body potential between a ground-state alkali atom (spherically symmetric) and one atoms of the solid matrix in the following form (the same is valid in the case of Li-(p-H
2) we will consider successively)
where
is the attractive two-body van der Waals potential, and
is a repulsive exchange or Coulomb potential, acting at short distances when a significant wavefunction’s overlap occurs. Several forms of these potentials have been proposed and used in the literature. We now introduce some relevant features of three of them, that we will use in the theoretical analysis given in the next subsection.
Taking the usual
dipolar attractive van der Waals potential energy for ground state systems, and a simple exponential for the repulsive short-distance component, we have the Buckingham–Hill potential [
36,
38,
39]
where the three parameters
, i.e., van der Waals constant, strength and distance scale of the repulsive potential, respectively, appear.
We assume that the total potential felt by the alkali atom inside the solid matrix is just the sum of the two-body potentials due to the first neighbors molecules of the matrix, thus neglecting many-body effects. In the Buckingham–Hill potential, we also neglect higher multipolar dispersion (long-range) interactions [
44], scaling with the distance as
and higher inverse powers of the distance [
46,
47]. These approximations are justified by the relatively large reticular distance,
for Ar and
for Ne [
11,
48,
49,
50].
For our purposes, it is important to relate the constants in the Buckingham–Hill potential (
4) to tabulated experimentally measured or theoretically computed quantities, such as the dimer equilibrium distance and dissociation energy (that is the potential energy at the equilibrium position). The van der Waals constant
is tabulated for many atoms and molecules in their ground or in excited states [
47,
51].
In order to evaluate the constant
A in (
4) in terms of known parameters, we can impose that
for
,
being the equilibrium distance of the alkali-inert-atom dimer. We immediately find
yielding
By taking into account that the quantities experimentally measured are the van der Waals constant
, the equilibrium distance
, and the dissociation energy
(that is the potential energy at the equilibrium distance), from (
6) we can obtain the following expression for
The complete expression of the potential is then
Thus, using Equation (
8), the Buckingham–Hill potential is entirely expressed in terms of hopefully known and tabulated parameters (for example, the various parameters for several alkali and RG atoms can be found in [
47,
51,
52]).
The Buckingham–Hill potential is a good approximation for distances around
, yielding a good approximation to the equilibrium distance, and for larger distances, where it correctly reduces to the van der Waals potential, but not for shorter distances: in fact, for distances smaller than
the Buckingham–Hill potential decreases to negative values, where it should grow and give the short-distance repulsion. In other words, the equilibrium position of the potential (
6) is an unstable point.
We thus need an appropriate approximation to the potential energy valid for distances smaller than the equilibrium distance. As will emerge in the next subsection, the short-distance case is what we are mainly interested to. In the short-distance region, that is, for distances smaller than the dimer’s equilibrium distance, we can use the Morse potential, given by [
36,
37]
being the dissociation energy of the dimer, that is the energy at the equilibrium distance
,
, with
the curvature of the potential at the equilibrium distance;
is a second parameter characterizing the interatomic potential energy.
Although the Morse potential (
9) is a good approximation to the short-range exchange/Coulomb potential, it fails at large distances. The Buckingham–Hill and the Morse potential are both expected to give a reasonably good approximation around the equilibrium distance.
The Morse potential (
9) contains three free parameters, but its mathematical expression does not allow to obtain a relation between them exploiting that its derivative must vanish at the equilibrium distance
. For this reason, since the constant
(that is the curvature of the potential at the equilibrium distance) is not always available from tabulated data in the literature, we suggest that in such cases we can resort to the Buckingham–Hill potential for its estimation. This could be done by calculating the modulus of the second derivative of the Buckingham–Hill potential at the equilibrium distance, and finally obtain the parameter
in the Morse potential (
9). We stress that some care should be used in this procedure, because, at the equilibrium distance, the BH potential has a maximum while the Morse potential has a minimum. Thus, using in the Morse potential the value of
obtained from the BH potential assumes that, around the equilibrium distance, the modulus of the curvature of both potentials are close to each other. Assuming this, from (
8) we obtain
Because in our case the distance between the alkali atom and any inert-gas atom molecule of the solid matrix is smaller than the equilibrium distance (, to be compared to , for the Rb-Ar dimer, for example), we expect that the Morse potential gives a better approximation to the true potential than the Buckingham–Hill potential. How good this procedure is for obtaining the parameter can be checked in the cases where is known, experimentally or theoretically, from independent considerations, as we will explicitly discuss later on for Rubidium atoms in a solid Argon or Neon matrix.
A third potential, accurate for the Lithium-parahydrogen dimer and that we will use in the next subsection for this case, although it involves a larger number of parameters, has been discussed and used in [
40,
41]. This potential has the following expression
with
where
,
,
,
are parameters relative to the atomic and molecular species considered, while
,
and
are the relative van der Waals constants. This potential has a spherical symmetry, because non-spherical components, depending from the specific geometrical configuration of the Li-H
2 system, are small; the potential (
11) is accurate in the distance range
2–12 Å. It has been also used for the H
2-H
2 and H
2-D
2 interaction energies [
40,
41].
3.2. Numerical Estimates of the Atoms-Matrix Interaction Energies
We can now use the potentials of
Section 3.1 to obtain approximated estimates of the value of the interaction energy between an alkali atom (Lithium, Sodium, Potassium, Rubidium) and an atom or molecule of the solid matrix (in this section we will consider Rubidium in Argon and Neon, and then Lithium in parahydrogen), assuming that the alkali atom replaces one atom or molecule of the solid crystal. From that, the interaction energy between a guest alkali atom and the solid RG or parahydrogen matrix can be easily obtained, assuming only pairwise interactions between the nearest neighbours. As mentioned before, this should be a fairly good approximation due to the large reticular distance of the solid matrix.
Possible trapping sites of alkali atoms in RG crystals, and their stability, have been recently studied in the literature [
53,
54,
55].
We first consider the Rb-Ar case and then the Rb-Ne case, comparing our theoretical estimates with the measured spectra reported in the previous section. At the end, we will also analyze the Li-(p-H2) case. Throughout this subsection, the units used are eV for the energy and Å for the distance.
Solid Argon and solid Neon have both a face-centered cubic (fcc) crystal structure; the reticular distance is
for Ar and
for Ne [
11,
48,
49,
50]. The guest alkali atom considered is Rubidium (Rb), as in the measurements reported in the previous section. We assume that the Rubidium atom substitutes one of the atoms of the matrix. Taking into account the fcc reticular structure of Argon and Neon, each Rubidium atom interacts with 12 atoms of the crystal at a distance
,
a being the reticular distance.
In order to evaluate the potential energy through the Morse or the Buckingham–Hill potential, we need the numerical values of the equilibrium distance
, the dissociation energy
, the curvature
of the potential energy at the equilibrium distance or the van der Waals constant
. Theoretical estimates of the equilibrium distance
and the dissociation energy
D can be found in several works [
46,
56,
57,
58]. The curvature of the potential energy at the equilibrium distance can be found in [
56,
57]. The
coefficients for several alkali atoms and RG atoms, both in the ground state and in excited levels, can be found in [
47,
51,
59,
60]. Although there is a good agreement in the literature on the values of
and
, the tabulated values of
D and
for Rb-Ne can significantly differ in different papers, and even in the same paper when different methods of calculation are used; the available data for Rb-Ar are instead much more uniform. For this reason we will concentrate mainly on the Rb-Ar case. We have chosen to use the data reported in [
57], where the three necessary parameters
,
D,
are calculated. We will also compare these data with those reported in [
58]. However, the small variability of the tabulated data for Rb-Ar is not much relevant for our purposes, because we only need an order-of-magnitude estimate of the interaction energy, and not its exact value.
- (i)
Rb-Ar
We first consider the ground-state configuration
of the Rb-Ar dimer; it corresponds to the electronic configuration of both the Rubidium and Argon atoms in the ground state. The relevant data are [
57]
For the same configuration, the data in [
58] for the equilibrium distance and the dissociation energy (the curvature of the potential is not explicitly given) are not very dissimilar:
,
.
As mentioned above, we need to evaluate the potential energy at the distance
, assuming that the Rubidium atom replaces one Argon atom of the matrix. Since
, we use the Morse potential (
9). The
parameter in Equation (
9) in the present case is therefore
. Using all these data in (
9), we immediately find
In this case we have been able to estimate the potential energy using the Morse potential only, because the potential curvature
is known. In
Section 3.1 we proposed that
could be obtained using the Buckingham–Hill potential through Equation (
10). The van der Waals constant
can be found in [
46,
47], for example, as
. From Equation (
10) we thus obtain
, that is relatively close to the tabulated value shown in (
13). This is an important consistency check for the analytical method presented in this section.
Summing the potential (
14) over the 12 nearest-neighbors of the Rubidium atom (pairwise summation), we obtain
Using the very recent data for the equilibrium distance and the dissociation energy in [
61],
,
, and the data for the curvature of the potential at the equilibrium distance communicated to us by the authors of that paper [
62],
, we get
, that is close to (
14). Interpolation of recent ab initio numerical data for the potential energy [
62] (see also [
61]), yields a somehow lower value,
, in any case not far from our analytical estimate. We can thus say that, even using numerical data from different sources, we obtain essentially the same approximate estimate of
.
Even if we are mainly interested to the ground state, as discussed later on, we can use the same procedure to estimate the potential energy of an excited state of the Rubidium atom in the solid Argon matrix. Specifically, we consider the first excited state in the Rb-Ar molecular configuration
(corresponding to a
configuration of Rubidium, and the Argon in its ground state). We use the following tabulated data:
,
[
58];
[
47]. Because in this case the distance at which we should evaluate the potential,
, is very close (slightly larger, to be precise) to the equilibrium distance
, we can use the Buckingham–Hill potential (
8), and we obtain
. Since the interatomic distance now considered is close to the equilibrium distance, we must expect that the Morse potential should yield the same result too. An explicit evaluation by using Equation (
9), with the potential curvature obtained from (
10), gives indeed practically the same result. After pairwise sum over the 12 nearest neighbors Argon atoms, we have
. These interaction energies are of the same order of magnitude of the ground-state interaction energies evaluated above.
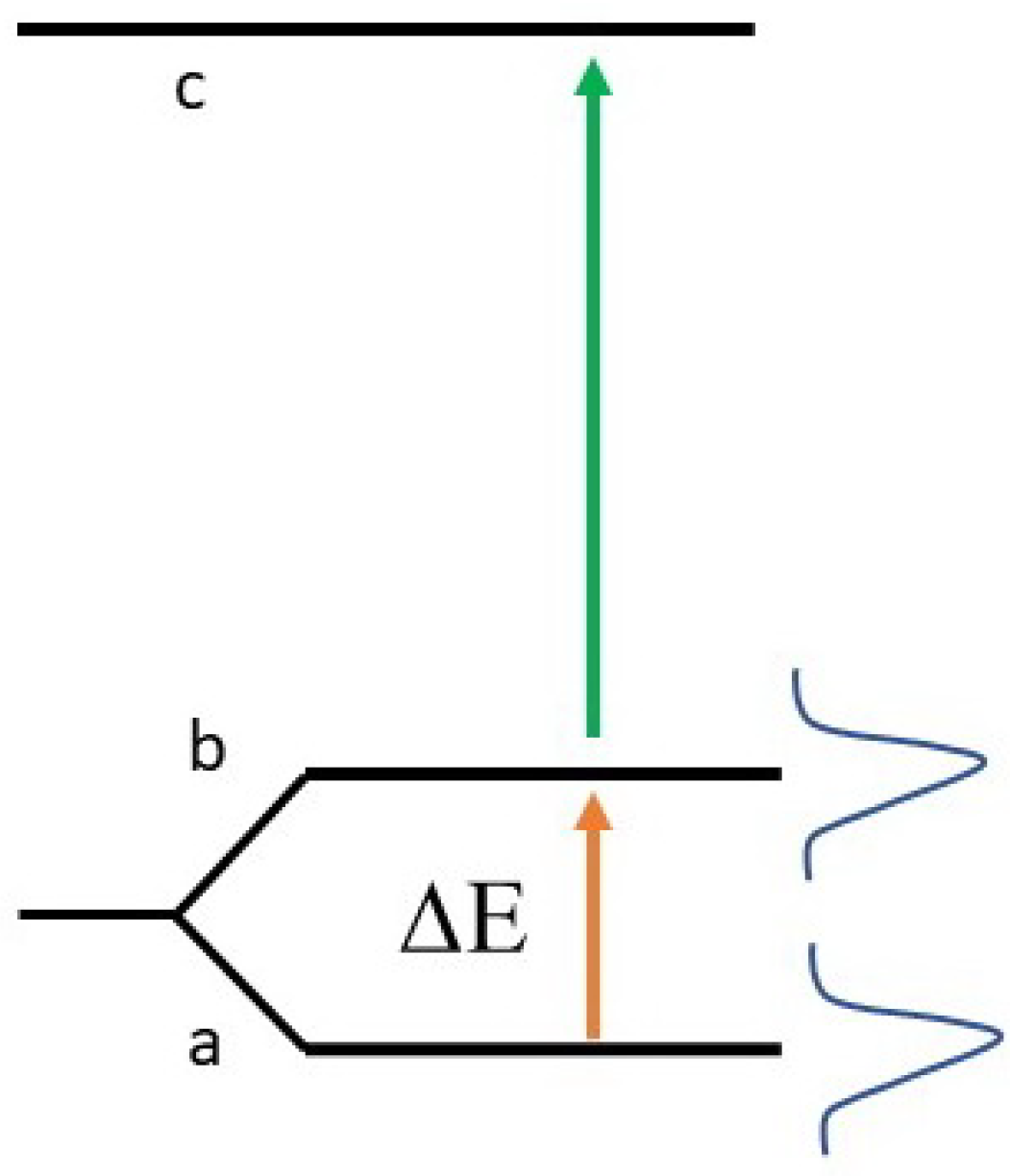
Our main aim here is the role of these interactions in the linewidth broadening of the guest atoms in the solid matrix. Together with other effects, phonon excitations in the matrix for example, they have also other consequences, of course, for example energy shifts due to the degeneracy lifting of excited atomic levels. We will not consider these effects in the present preliminary study. We will limit our considerations only to the contribution of the interactions of the doping atoms with the matrix for their ground state, splitted in the two Zeeman sublevels by the external magnetic field. With reference to the detection scheme of
Figure 1, this is reasonable if
c represents the edge of the crystal conduction band (the axion transition
does not involve a change in the atomic wavefunction, and thus it should not perturb the crystal structure). If
c in
Figure 1 represents an excited level of the doping atoms, we should also add the linewidth of the excited level and the possible perturbation (phonons, for example) due to the change of the atomic wavefunction in the transition between level
a or
b and the excited level
c. For this reason, a detailed comparison of the experimental spectral widths reported in the previous section with our theoretical analysis of the interaction energies between the ground-state guest atom with the cold matrix is not immediate; however, the present theoretical analysis of this contribution is significant since it can give useful hints on how to minimize this contribution to the width of the ground state, which is the level from which the axion absorption occurs.
We now assume that the contribution to the inhomogeneous broadening of the lines due to the interactions between the dilute alkali atoms and the RG solid matrix is some small fraction of the energy given in (
15), plus eventually that relative to the excited level in the case of a transition between two discrete levels, that we have seen to be of the same order of magnitude for the Rb-Ar system. Which value has this fraction is very difficult to evaluate from a theoretical point of view only, because it depends on specific details of the position distribution of the alkali atoms inside the solid matrix, for example the inhomogeneity of their position, imperfections of the crystal structures, etc. Thus, it can significantly depend on how the solid matrix is grown, and, importantly, how the dilute alkali atoms are diffused inside the matrix. We can assume that the main effect is due to a (slight) inhomogeneous distribution of the position of the guest alkali atoms in the RG solid matrix. Comparing our experimental results for the spectral linewidth, reported in
Section 2, with our present theoretical prediction of the atom–matrix interaction energy, we now show that we can predict a semi-quantitative estimate of the atoms’ position distribution in the matrix. This analysis is also based on the fact, known in the literature and mentioned in the previous section, that each triplet of the spectrum is relative to doping atoms in the same trapping site [
35].
Quantitatively, a spreading of the alkali positions inside the crystal determines an inhomogeneous contribution to the linewidth of the spectrum, which strongly depends on how steep the potential energy is at their average position, of course. Assuming, as before, that the alkali atoms replace one of the atoms of the matrix, being
, we can expand the Morse potential (
9) around
(taking into account the measured relative energy widths given in (
2), we expect a relatively sharp distribution of the positions). We indicate with
a small displacement from the average distance
, which is equal to
in the present Rb-Ar case, we can obtain the consequent change of the potential energy as
and thus the relative absolute value of the change of the potential energy is
This relation allows us to estimate the position dispersion
of the alkali atoms in the solid matrix, once the relative dispersion of the potential energy is known. The latter can be obtained from the spectral experimental results of the previous section, specifically from the relative energy widths (
2). In the case of Rb-Ar here considered, for the peak 3 of
Table 1 (see also
Figure 3) with an energy witdh of
, and using the numerical data in (
13) and (
17) yields the Argon-atoms position dispersion
that means a relative dispersion of ∼0.5%. Indeed, this should be considered as an upper limit to the position dispersion around the trapping site, because other effects, not considered here, can contribute to the line broadening.
Our experimental measurements, combined with our theoretical model, have thus allowed us to obtain relevant hints on how the guest atoms in our Rb-Ar experimental setup diffuse in the cold matrix, and relate the measured spectral widths to the alkali–matrix interactions. This method can be also applied also to other doping-atoms matrix, for example Rubidium atoms in a cold Neon matrix.
- (ii)
Rb-Ne
We now consider the Rb-Ne case, whose spectral measurements have been reported at the end of
Section 2. The relevant numerical values for equilibrium distance, dissociation energy and van der Waals constant can be found in [
46,
47,
56,
57,
61,
63]. In this case, some differences are found in the literature among the data obtained with different methods, in particular for the dissociation energy. This can yield different estimates of the interaction energy in the solid matrix, depending on the numerical data used.
Solid Neon has a fcc crystal structure as solid Argon, with a reticular distance
. We first use the very recent data of the equilibrium distance and dissociation energy for the Rb-Ne dimer reported in [
61],
and
, respectively, supplemented with the value of the potential curvature at the potential minimum [
62],
. In this case we obtain
which is larger than that for Rb-Ar. A similar result is obtained using the data in [
57],
,
,
, where the value of the curvature
of the potential at the equilibrium position has been inferred from their data of the harmonic wavenumbers, obtaining in this case
. This is also confirmed by other recent numerical data [
62] (see also [
61]), yielding
: this value is fairly lower than our theoretical estimate based on the Morse potential (it is, however, larger compared to the Rb-Ar case), showing again a larger variability of the energies in the Rb-Ne case according to the analytical or numerical method used.
Although our theoretically predicted values for the ground-state interaction energy for Rb-Ne are larger than for Rb-Ar, as a comparison with (
14) immediately shows, while our spectra of
Section 2 show sharper linewidths in the Rb-Ne case, this is not necessarily a contradiction between experimental results and theoretical analysis. In fact, also other aspects should be taken into account for a direct comparison of the linewidths. Firstly, our theoretical estimates involve only the ground state, while the spectral linewidths of
Section 2 include also the effects of the excited level, and consequent rearrangement of the wavefunction distribution and possible phonon emission. A wavefunction rearrangement is present also in the considered case of transition to the crystal conduction band, but we expect its effect to be much smaller with respect to the transition to a higher excited level, which involves a larger size of the final discrete state and thus a larger perturbation of the crystal. Thus, we guess that our ground-state linewidth estimates could be directly compared to future measurements of transitions of the doping atom’s electron from the ground state (also if Zeeman shifted) to the crystal conduction band. Secondly, we cannot exclude that the position distribution of the guest atoms in the solid matrix is smaller in Neon compared to Argon. Indeed, the two solid matrices have different physical properties, related to the interactions between the matrix atoms, for example the condensation temperature,
for Ar and
for Ne, and the reticular distance: thus, nontrivial differences in the way the guest atoms arrange themselves in the matrix are not unexpected. We plan to discuss these points in a future work. In any case, we believe that the case of Neon deserves a more precise analysis from the theoretical side, also because the alkali-RG atoms distance in the Neon matrix is smaller than in the Argon matrix, in particular in comparison with the corresponding equilibrium distance (
,
for Rb-Ar;
,
for Rb-Ne). At shorter distances, the potential energy is very steep, as, consistently, the plots of the potential energies in [
61] clearly show. All this makes the details of the model more critical: even a small variation of the relevant parameters can yield a consistent variation of the estimated potential energy.
- (iii)
Li-(p-H2)
Finally, we consider theoretically the Li-(p-H2) case; that is, dilute Lithium atoms diffused in a solid parahydrogen matrix. We will obtain a strong indication that this system shows a quite smaller guest-atom–matrix interaction energy.
The p-H
2 molecule has nuclear spin
and, at low temperatures, an even total angular momentum
J, with a spherical symmetry yielding vanishing permanent multipolar momenta [
64]. Due to the spherical symmetry of the parahydrogen molecule, considerations on the potentials similar to the rare gases, discussed above, can be applied. Solid parahydrogen has a Hexagonal closed packed (Hcp) structure, with a quite large intermolecular distance compared to the relevant atomic or molecular dimensions. We assume that the alkali atom is placed at the hexagon center, substituting one of the H
2 molecules, as shown in
Figure 6; due to the large intermolecular distance of the molecules in the p-H
2 matrix, its presence should not significantly change the structure of the crystal. Furthermore, the known large compressibility of parahydrogen solids allows us to easily accommodate the alkali-atom impurity [
65]. The reticular distance for the parahydrogen crystal is
[
2,
66].
The relevant tabulated data for the dissociation energy
D and the equilibrium distance
can be found in [
52], while the van der Waals constant
can be found in [
51]; the tabulated values are
,
,
. From (
7) we then get
. Since the radius of the Lithium atom,
, is quite smaller than the alkali–parahydrogen distance in the solid matrix,
, our pairwise-summation of the potentials between nearest-neighbors pairs, as well as neglecting higher multipolar dispersion interactions, should be reasonably good approximations. Since the Li-H
2 distance in the solid matrix is smaller than the equilibrium distance,
, we expect that the Morse potential should give a better approximation to the true potential than the Buckingham–Hill potential.
With the numerical values above, taking the second derivative of the Buckingham–Hill potential (
8) and (Equation (
10)), and evaluating it for the distance
between the alkali atom and any of the nearest-neighbors parahydrogen molecules (see
Figure 6), we obtain
, and then
, to be used in the Morse potential (
9).
Taking into account that each Lithium atom has six nearest neighbors parahydrogen molecules of the solid matrix, and assuming a pairwise summation, we obtain the total interaction energy of each Lithium atom in the parahydrogen solid matrix
We can also calculate the Li-(p-H
2) interaction energy using the potential (
11), with the following parameters given in [
41]; in atomic units, for the
configuration (ground state), they are:
,
,
,
,
,
,
[
41]. We obtain
This value is about one half of that predicted from the Morse potential, and so it is even more favorable for the feasibility of our proposed detection scheme. In fact, after summation over the six nearest-neighbors parahydrogen molecules, it yields a total potential energy of the order of one meV.
Comparing (
20) and (
22) with (
14) and (
19), we see that the interaction energy in the present case is rather smaller than for both Rb-Ar and Rb-Ne. On the basis of our previous considerations, this should give a rather smaller energy width of the Lithium ground state in the matrix, at least for the contribution from its interaction with the lattice molecules, and assuming a similar dispersion of the positions of the alkali atoms in the cold solid matrix. Some experimental spectroscopic data on the Li-(p-H
2) system can be found in [
67], while theoretical aspects relevant for the absorption spectra are given in [
41]. In [
67], the spectral linewidth of the Lithium atoms in the p-H
2 cold matrix are of the order of 100 meV, but they cannot be directly compared with the ground-state interaction energies here evaluated. In fact, spectral lines, as already mentioned for the Ar and Ne cases, involve also other effects, such as, for example, the width of the excited level, energy spitting of degenerate excited levels due to crystal field, as well as phonon excitation in the crystal, that could be relevant since parahydrogen is a quantum crystal. We however expect that these effects play a less relevant role in our case, when we consider the transition between the alkali–atom Zeeman sublevel
b (reached after the assumed axion absorption) to the crystal conduction band, whose edge in
Figure 1 is indicated by
c. The reason is that in this case, the atomic wavefunction of the sublevels
a and
b is the same, and there are not other discrete levels involved in the transition.
Finally, taking into account the theoretical results for Li-(p-H
2) of this section, we expect that the Lithium-parahydrogen system could be a very promising physical system for the proposed axion detecting scheme, exploiting the transitions in
Figure 1, with
c indicating the edge of the conduction band of the cold crystal.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}