
DECIDE: A Deterministic Mixed Quantum-Classical Dynamics Approach




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Wigner-Weyl Transform
3. DECIDE Mixed Quantum-Classical Dynamics
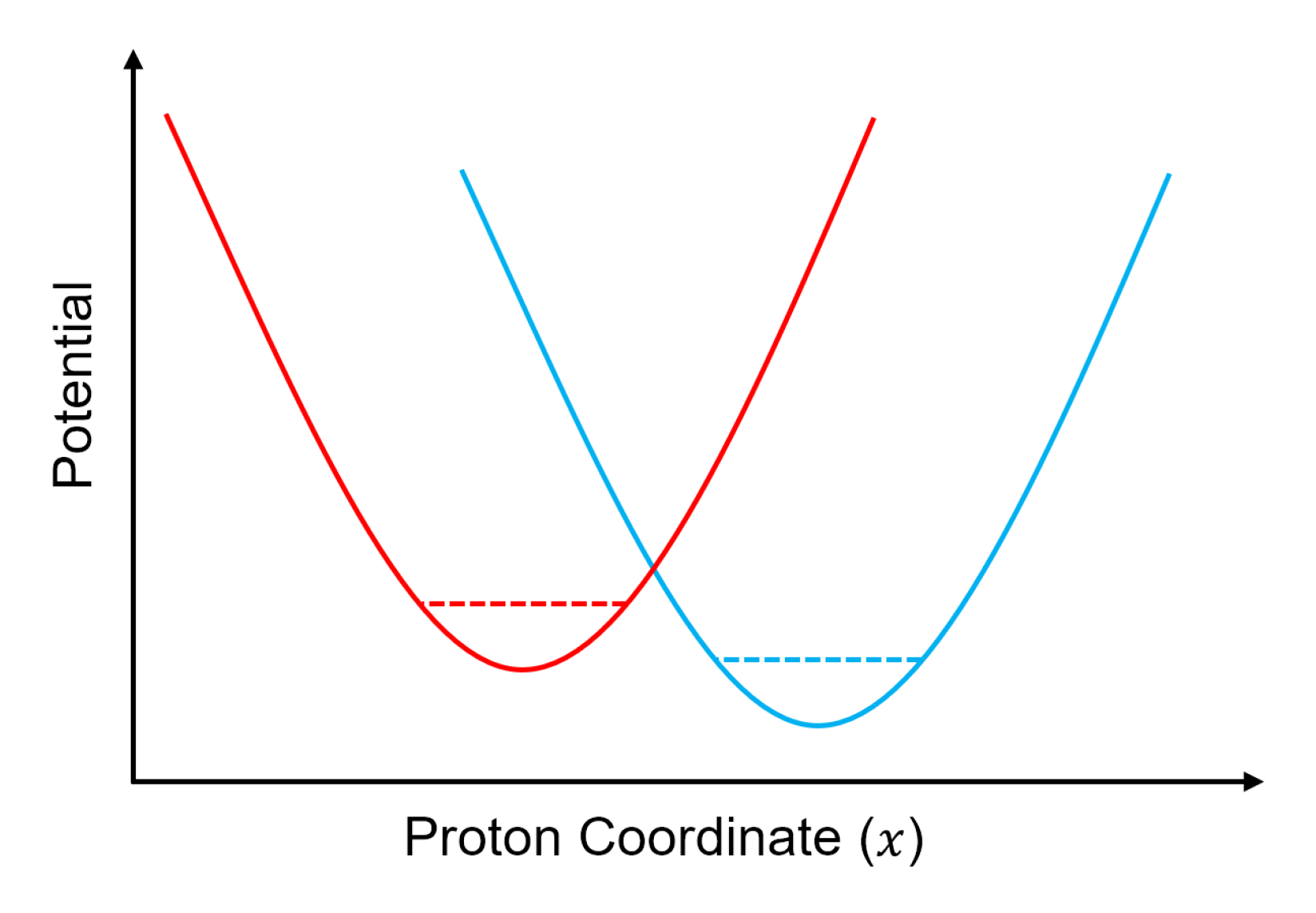
4. Hydrogen Bond Model
4.1. Model
4.2. Dynamics Simulation Details
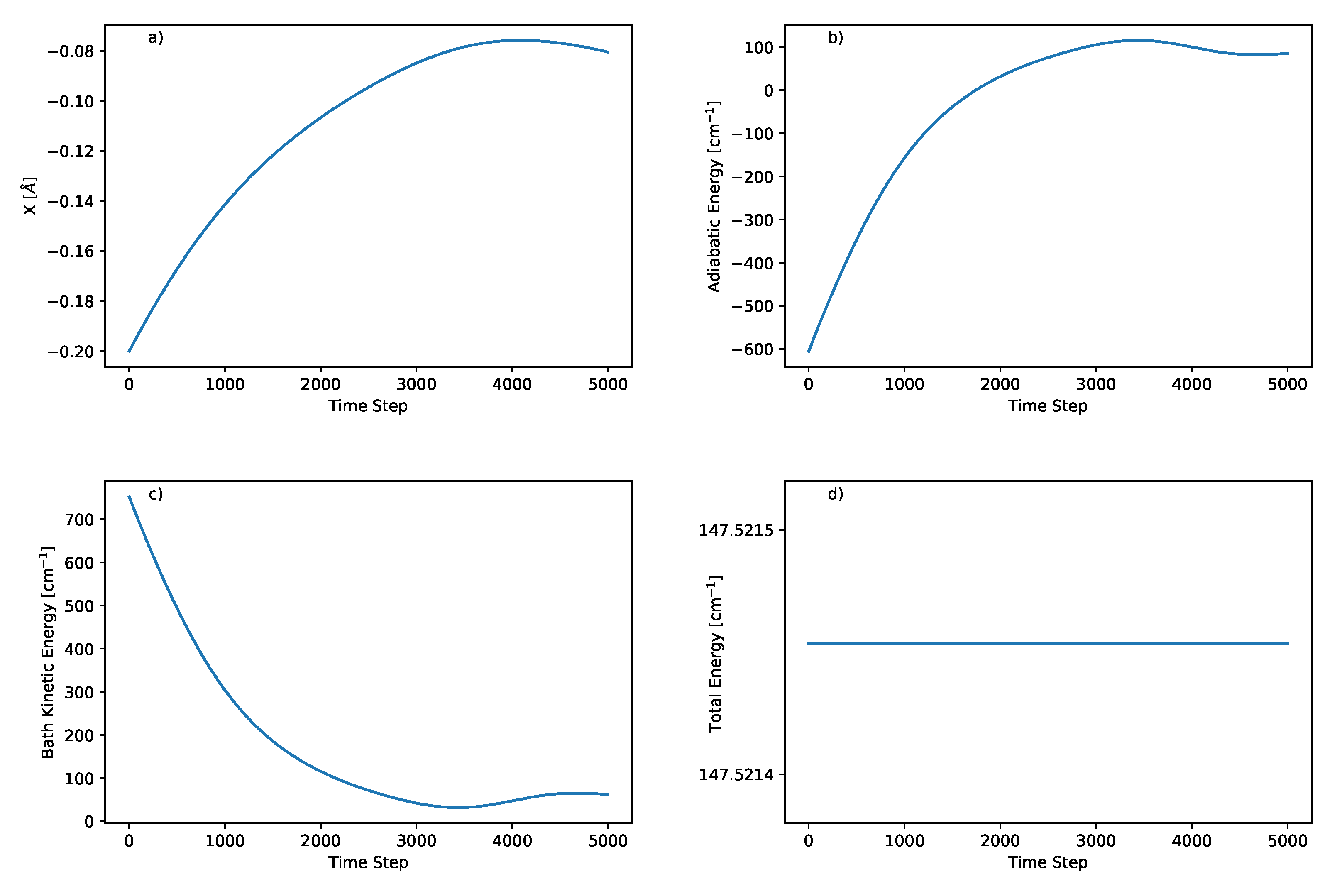
4.3. Results
4.4. Limitations of Position Representation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DECIDE | Deterministic evolution of coordinates with initial decoupled equations |
| DOF | Degrees of freedom |
| QCLE | Quantum-classical Liouville equation |
| 1D | One-dimensional |
References
- Tully, J.C. Mixed quantum-classical dynamics. Faraday Discuss. 1998, 110, 407–419. [Google Scholar] [CrossRef]
- Kapral, R.; Ciccotti, G. Mixed quantum-classical dynamics. J. Chem. Phys. 1999, 110, 8919–8929. [Google Scholar] [CrossRef]
- Kapral, R. Progress in the theory of mixed quantum-classical dynamics. Annu. Rev. Phys. Chem. 2006, 57, 129–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergi, A.; Hanna, G.; Grimaudo, R.; Messina, A. Quasi-Lie brackets and the breaking of time-translation symmetry for quantum systems embedded in classical baths. Symmetry 2018, 10, 518. [Google Scholar] [CrossRef] [Green Version]
- Sergi, A. Embedding quantum systems with a non-conserved probability in classical environments. Theor. Chem. Accounts 2015, 134, 79. [Google Scholar] [CrossRef] [Green Version]
- Hanna, G.; Kapral, R. Quantum-classical Liouville dynamics of nonadiabatic proton transfer. J. Chem. Phys. 2005, 122, 244505. [Google Scholar] [CrossRef]
- Hammes-Schiffer, S.; Tully, J.C. Proton transfer in solution: Molecular dynamics with quantum transitions. J. Chem. Phys. 1994, 101, 4657–4667. [Google Scholar] [CrossRef] [Green Version]
- Rekik, N.; Hsieh, C.Y.; Freedman, H.; Hanna, G. A mixed quantum-classical Liouville study of the population dynamics in a model photo-induced condensed phase electron transfer reaction. J. Chem. Phys. 2013, 138, 144106. [Google Scholar] [CrossRef]
- Shakib, F.A.; Hanna, G. New insights into the nonadiabatic state population dynamics of model proton-coupled electron transfer reactions from the mixed quantum-classical Liouville approach. J. Chem. Phys. 2016, 144, 024110. [Google Scholar] [CrossRef]
- Fang, J.Y.; Hammes-Schiffer, S. Proton-coupled electron transfer reactions in solution: Molecular dynamics with quantum transitions for model systems. J. Chem. Phys. 1997, 106, 8442–8454. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.Y.; Hammes-Schiffer, S. Excited state dynamics with nonadiabatic transitions for model photoinduced proton-coupled electron transfer reactions. J. Chem. Phys. 1997, 107, 5727–5739. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.Y.; Hammes-Schiffer, S. Nonadiabatic dynamics for processes involving multiple avoided curve crossings: Double proton transfer and proton-coupled electron transfer reactions. J. Chem. Phys. 1997, 107, 8933–8939. [Google Scholar] [CrossRef] [Green Version]
- Soudackov, A.V.; Hammes-Schiffer, S. Removal of the double adiabatic approximation for proton-coupled electron transfer reactions in solution. Chem. Phys. Lett. 1999, 299, 503–510. [Google Scholar] [CrossRef]
- Shakib, F.A.; Hanna, G. An analysis of model proton-coupled electron transfer reactions via the mixed quantum-classical Liouville approach. J. Chem. Phys. 2014, 141, 044122. [Google Scholar] [CrossRef]
- Shakib, F.; Hanna, G. Mixed quantum-classical Liouville approach for calculating proton-coupled electron-transfer rate constants. J. Chem. Theory Comput. 2016, 12, 3020–3029. [Google Scholar] [CrossRef]
- Sayfutyarova, E.R.; Goings, J.J.; Hammes-Schiffer, S. Electron-coupled double proton transfer in the Slr1694 BLUF photoreceptor: A multireference electronic structure study. J. Phys. Chem. B 2018, 123, 439–447. [Google Scholar] [CrossRef]
- Panitchayangkoon, G.; Voronine, D.V.; Abramavicius, D.; Caram, J.R.; Lewis, N.H.; Mukamel, S.; Engel, G.S. Direct evidence of quantum transport in photosynthetic light-harvesting complexes. Proc. Natl. Acad. Sci. USA 2011, 108, 20908–20912. [Google Scholar] [CrossRef] [Green Version]
- Kelly, A.; Rhee, Y.M. Mixed quantum-classical description of excitation energy transfer in a model Fenna-Matthews-Olsen complex. J. Phys. Chem. Lett. 2011, 2, 808–812. [Google Scholar] [CrossRef]
- Harush, E.Z.; Dubi, Y. Do photosynthetic complexes use quantum coherence to increase their efficiency? Probably not. Sci. Adv. 2021, 7, eabc4631. [Google Scholar] [CrossRef]
- Segal, D.; Agarwalla, B.K. Vibrational heat transport in molecular junctions. Annu. Rev. Phys. Chem. 2016, 67, 185–209. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Hsieh, C.Y.; Segal, D.; Hanna, G. Heat transfer statistics in mixed quantum-classical systems. J. Chem. Phys. 2018, 149, 224104. [Google Scholar] [CrossRef]
- Carpio-Martínez, P.; Hanna, G. Nonequilibrium heat transport in a molecular junction: A mixed quantum-classical approach. J. Chem. Phys. 2019, 151, 074112. [Google Scholar] [CrossRef]
- Kelly, A. Mean field theory of thermal energy transport in molecular junctions. J. Chem. Phys. 2019, 150, 204107. [Google Scholar] [CrossRef]
- Grimaldi, A.; Sergi, A.; Messina, A. Evolution of a non-Hermitian quantum single-molecule junction at constant temperature. Entropy 2021, 23, 147. [Google Scholar] [CrossRef]
- Uken, D.A.; Sergi, A. Quantum dynamics of a plasmonic meta-molecule with a time-dependent driving. Theor. Chem. Acc. 2015, 134, 141. [Google Scholar] [CrossRef] [Green Version]
- Sergi, A. Communication: Quantum dynamics in classical spin baths. J. Chem. Phys. 2013, 139, 031101. [Google Scholar] [CrossRef] [Green Version]
- Sergi, A. Computer simulation of quantum dynamics in a classical spin environment. Theor. Chem. Acc. 2014, 133, 1495. [Google Scholar] [CrossRef] [Green Version]
- Aleksandrov, I.V. The statistical dynamics of a system consisting of a classical and a quantum subsystem. Z. Naturforsch. A 1981, 36, 902–908. [Google Scholar] [CrossRef]
- Gerasimenko, V.I. Dynamical equations of quantum-classical systems. Theor. Math. Phys. 1982, 50, 77–87. [Google Scholar] [CrossRef]
- Kapral, R. Quantum dynamics in open quantum-classical systems. J. Phys. Condens. Matter 2015, 27, 073201. [Google Scholar] [CrossRef] [Green Version]
- MacKernan, D.; Kapral, R.; Ciccotti, G. Sequential short-time propagation of quantum-classical dynamics. J. Phys. Condens. Matter 2002, 14, 9069–9076. [Google Scholar] [CrossRef]
- Mac Kernan, D.; Ciccotti, G.; Kapral, R. Trotter-based simulation of quantum-classical dynamics. J. Phys. Chem. B 2008, 112, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Uken, D.A.; Sergi, A.; Petruccione, F. Filtering schemes in the quantum-classical Liouville approach to non-adiabatic dynamics. Phys. Rev. E 2013, 88, 033301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapral, R. Surface hopping from the perspective of quantum-classical Liouville dynamics. Chem. Phys. 2016, 481, 77–83. [Google Scholar] [CrossRef] [Green Version]
- Dell’Angelo, D.; Hanna, G. Self-consistent filtering scheme for efficient calculations of observables via the mixed quantum-classical Liouville approach. J. Chem. Theory Comput. 2016, 12, 477–485. [Google Scholar] [CrossRef]
- Hanna, G.; Sergi, A. Simulating quantum dynamics in classical nanoscale environments. In Theoretical Chemistry for Advanced Nanomaterials; Springer: Singapore, 2020; pp. 515–544. [Google Scholar]
- Nassimi, A.; Bonella, S.; Kapral, R. Analysis of the quantum-classical Liouville equation in the mapping basis. J. Chem. Phys. 2010, 133, 134115. [Google Scholar] [CrossRef] [Green Version]
- Kelly, A.; van Zon, R.; Schofield, J.; Kapral, R. Mapping quantum-classical Liouville equation: Projectors and trajectories. J. Chem. Phys. 2012, 136, 084101. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.W.; Rhee, Y.M. Improving long time behavior of Poisson bracket mapping equation: A non-Hamiltonian approach. J. Chem. Phys. 2014, 140, 184106. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Hanna, G. Efficient and deterministic propagation of mixed quantum-classical Liouville dynamics. J. Phys. Chem. Lett. 2018, 9, 3928–3933. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Segal, D.; Hanna, G. Hybrid quantum-classical simulation of quantum speed limits in open quantum systems. J. Phys. A Math. Theor. 2019, 52, 215301. [Google Scholar] [CrossRef] [Green Version]
- Carpio-Martínez, P.; Hanna, G. Quantum bath effects on nonequilibrium heat transport in model molecular junctions. J. Chem. Phys. 2021, 154, 094108. [Google Scholar] [CrossRef]
- Liu, J.; Segal, D.; Hanna, G. Loss-free excitonic quantum battery. J. Phys. Chem. C 2019, 123, 18303–18314. [Google Scholar] [CrossRef]
- Wigner, E.P. On the quantum correction for thermodynamic equilibrium. In Part I: Physical Chemistry. Part II: Solid State Physics; Springer: Berlin/Heidelberg, Germany, 1997; pp. 110–120. [Google Scholar]
- Case, W.B. Wigner functions and Weyl transforms for pedestrians. Am. J. Phys. 2008, 76, 937–946. [Google Scholar] [CrossRef] [Green Version]
- Imre, K.; Özizmir, E.; Rosenbaum, M.; Zweifel, P. Wigner method in quantum statistical mechanics. J. Math. Phys. 1967, 8, 1097–1108. [Google Scholar] [CrossRef] [Green Version]
- Moyal, J.E. Quantum mechanics as a statistical theory. In Mathematical Proceedings of the Cambridge Philosophical Society; Cambridge University Press: Cambridge, UK, 1949; Volume 45, pp. 99–124. [Google Scholar]
- Curtright, T.L.; Fairlie, D.B.; Zachos, C.K. A Concise Treatise on Quantum Mechanics in Phase Space; World Scientific Publishing Company: Singapore, 2013. [Google Scholar]
- Süli, E.; Mayers, D.F. An Introduction to Numerical Analysis; Cambridge University Press: Cambridge, UK, 2003. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Sergi, A.; Hanna, G. DECIDE: A Deterministic Mixed Quantum-Classical Dynamics Approach. Appl. Sci. 2022, 12, 7022. https://doi.org/10.3390/app12147022
Liu Z, Sergi A, Hanna G. DECIDE: A Deterministic Mixed Quantum-Classical Dynamics Approach. Applied Sciences. 2022; 12(14):7022. https://doi.org/10.3390/app12147022
Chicago/Turabian StyleLiu, Zhe, Alessandro Sergi, and Gabriel Hanna. 2022. "DECIDE: A Deterministic Mixed Quantum-Classical Dynamics Approach" Applied Sciences 12, no. 14: 7022. https://doi.org/10.3390/app12147022
APA StyleLiu, Z., Sergi, A., & Hanna, G. (2022). DECIDE: A Deterministic Mixed Quantum-Classical Dynamics Approach. Applied Sciences, 12(14), 7022. https://doi.org/10.3390/app12147022