
Nucleofection as an Efficient Method for Alpha TC1-6 Cell Line Transfection

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmids
2.3. Polyethylenimine (PEI)-Based Cell Transfection
Protocol for Chemical Transfection
- Preparatory work
- ○
- Two days before transfection, plate 1.5 × 106 into six-well plates in DMEM.
- ○
- Warm PBS, complete and incomplete DMEM (without FBS) to 37 °C.
- Mix plasmid DNA in incomplete DMEM media at a ratio of 2 µg DNA in 100 µL media for transfection in a six-well plate. Incubate 5 min at RT.
- Add 6 µL of PEI (1 mg/mL of working solution) per 100 µL of incomplete DMEM. Mix them by tapping and incubate for 5 min at RT.
- Add DNA to PEI drop-wise with constant tapping of the cuvette. Incubate for 20 min at RT.
- Add the final mix of DNA:PEI drop-wise to the cells with 1.8 mL complete DMEM.
- Gently rock the plate with the cells for uniform distribution of mix for transfection.
- Incubate at 37 °C for 16 h.
- Rinse the cells with PBS, and propagate cells until analysis.
2.4. Nucleofection
Optimized Protocol for Nucleofection for the αTC1-6 Cell Line
- After reaching 70% of cell confluency, aspirate the medium from the flask. Wash the cells once with PBS and harvest them by using cell dissociation buffer.
- Determine cell density.
- Centrifuge 5 × 106 cells per one 100 µL single Nucleocuvette™ at 90× g for 10 min at RT and completely remove the supernatant.
- Add 100 µL 4D-Nucleofector™ SF Solution with supplement and 7.5-µg plasmids on dry cells pellet. Mix gently with pipetting.
- Transfer cells into the 100-µL single Nucleocuvette™.
- Place Nucleocuvette™ into the retainer of the 4D-Nucleofector™ X Unit and start the CM-156 nucleofection program.
- After run completion, add 400 μL of RPMI medium to each Nucleocuvette™ and incubate them for 10 min at 37 °C.
- Gently resuspend cells by pipetting two to three times and transfer cells in pre-incubated six-well plates with complete DMEM.
- Propagate cells until analysis.
2.5. Fluorescent Microscopy
2.6. Flow Cytometry and Cell Sorting
2.7. RNA Isolation and Real-Time Quantitative PCR (RT-qPCR)
- Cas9: Fw 5′–TCAGGCGGCAAGAGGATTTC-3′, Rev 5′-AGTCATCCACGCGAATCTGG-3′,
- REEP 5: Fw 5′-TCATCGGACTGGTGGCTTTG-3′, Rev 5′-GTTGGGACTCTCGATGGCTT-3′.
2.8. Immunocytochemistry
2.9. Statistical Analysis
3. Results and Discussion
3.1. PEI-Based Transfection of αTC1-6 Cells
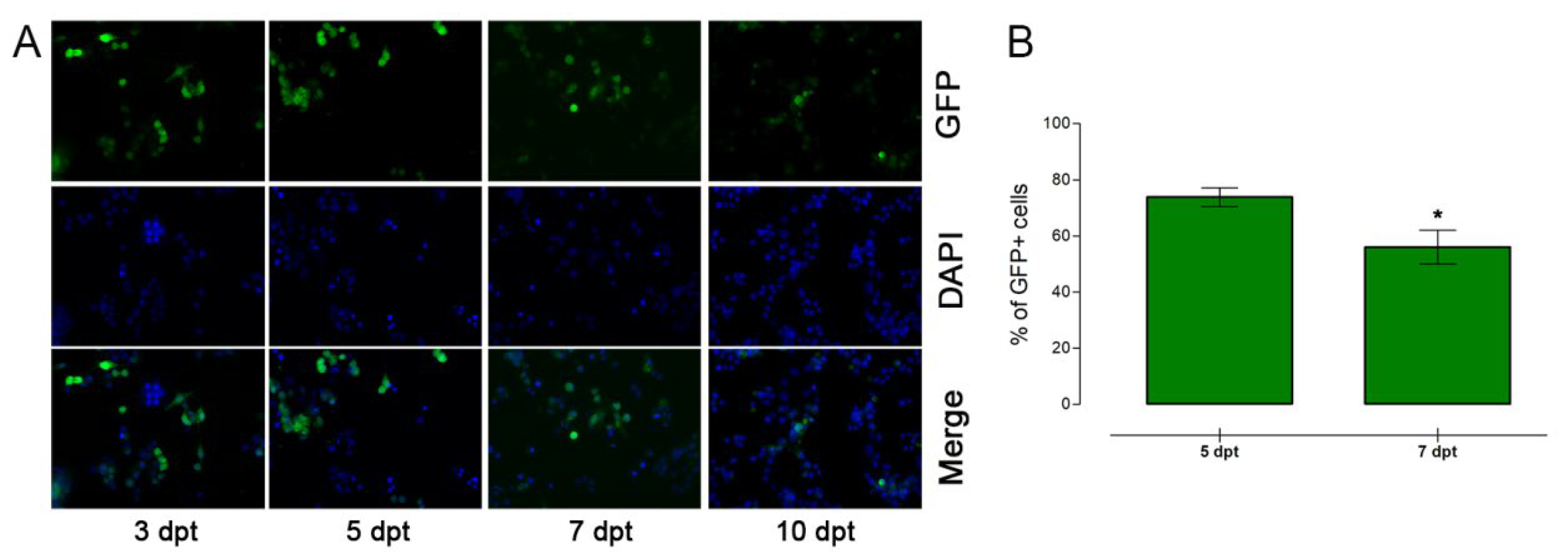
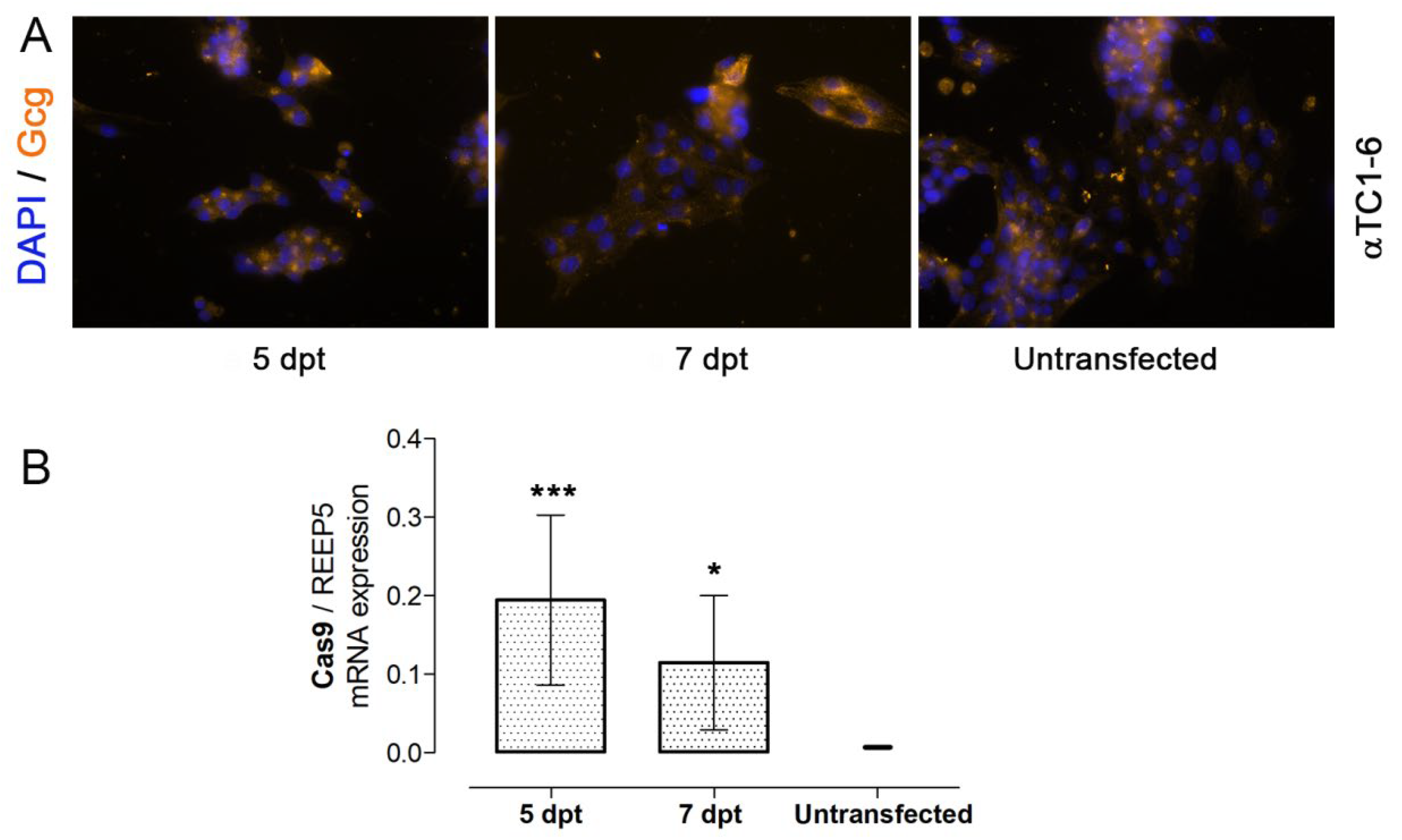
3.2. Nucleofection of αTC1-6 Cells
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stepper, P.; Kungulovski, G.; Jurkowska, R.Z.; Chandra, T.; Krueger, F.; Reinhardt, R.; Reik, W.; Jeltsch, A.; Jurkowski, T.P. Efficient targeted DNA methylation with chimeric dCas9–Dnmt3a–Dnmt3L methyltransferase. Nucleic Acids Res. 2016, 45, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Vojta, A.; Dobrinić, P.; Tadić, V.; Bočkor, L.; Korać, P.; Julg, B.; Klasić, M.; Zoldoš, V. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016, 44, 5615–5628. [Google Scholar] [CrossRef]
- Xie, N.; Zhou, Y.; Sun, Q.; Tang, B. Novel Epigenetic Techniques Provided by the CRISPR/Cas9 System. Stem Cells Int. 2018, 2018, 7834175. [Google Scholar] [CrossRef] [PubMed]
- Amabile, A.; Migliara, A.; Capasso, P.; Biffi, M.; Cittaro, D.; Naldini, L.; Lombardo, A. Inheritable Silencing of Endogenous Genes by Hit-and-Run Targeted Epigenetic Editing. Cell 2016, 167, 219–232.e214. [Google Scholar] [CrossRef]
- Stanojevic, V.; Habener, J.F. Evolving function and potential of pancreatic alpha cells. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 859–871. [Google Scholar] [CrossRef]
- Thorel, F.; Népote, V.; Avril, I.; Kohno, K.; Desgraz, R.; Chera, S.; Herrera, P.L. Conversion of adult pancreatic α-cells to β-cells after extreme β-cell loss. Nature 2010, 464, 1149–1154. [Google Scholar] [CrossRef] [PubMed]
- Thyssen, S.; Arany, E.; Hill, D.J. Ontogeny of Regeneration of β-Cells in the Neonatal Rat after Treatment with Streptozotocin. Endocrinology 2006, 147, 2346–2356. [Google Scholar] [CrossRef]
- Saleh, M.; Gittes, G.K.; Prasadan, K. Alpha-to-beta cell trans-differentiation for treatment of diabetes. Biochem. Soc. Trans. 2021, 49, 2539–2548. [Google Scholar] [CrossRef]
- Kim, T.K.; Eberwine, J.H. Mammalian cell transfection: The present and the future. Anal. Bioanal. Chem. 2010, 397, 3173–3178. [Google Scholar] [CrossRef]
- Chong, Z.X.; Yeap, S.K.; Ho, W.Y. Transfection types, methods and strategies: A technical review. PeerJ 2021, 9, e11165. [Google Scholar] [CrossRef]
- Han, X.; Fang, Q.; Yao, F.; Wang, X.; Wang, J.; Yang, S.; Shen, B.Q. The heterogeneous nature of polyethylenimine-DNA complex formation affects transient gene expression. Cytotechnology 2009, 60, 63. [Google Scholar] [CrossRef][Green Version]
- Zakeri, A.; Kouhbanani, M.A.J.; Beheshtkhoo, N.; Beigi, V.; Mousavi, S.M.; Hashemi, S.A.R.; Karimi Zade, A.; Amani, A.M.; Savardashtaki, A.; Mirzaei, E.; et al. Polyethylenimine-based nanocarriers in co-delivery of drug and gene: A developing horizon. Nano Rev. Exp. 2018, 9, 1488497. [Google Scholar] [CrossRef]
- Mehier-Humbert, S.; Guy, R.H. Physical methods for gene transfer: Improving the kinetics of gene delivery into cells. Adv. Drug Deliv. Rev. 2005, 57, 733–753. [Google Scholar] [CrossRef]
- Meng, L.; Liu, X.; Wang, Y.; Zhang, W.; Zhou, W.; Cai, F.; Li, F.; Wu, J.; Xu, L.; Niu, L.; et al. Sonoporation of Cells by a Parallel Stable Cavitation Microbubble Array. Adv. Sci. 2019, 6, 1900557. [Google Scholar] [CrossRef]
- Bessis, N.; GarciaCozar, F.J.; Boissier, M.C. Immune responses to gene therapy vectors: Influence on vector function and effector mechanisms. Gene Ther. 2004, 11, S10–S17. [Google Scholar] [CrossRef]
- Guo, X.; Wang, H.; Li, Y.; Leng, X.; Huang, W.; Ma, Y.; Xu, T.; Qi, X. Transfection reagent Lipofectamine triggers type I interferon signaling activation in macrophages. Immunol. Cell Biol. 2019, 97, 92–96. [Google Scholar] [CrossRef]
- Hohenstein, K.A.; Pyle, A.D.; Chern, J.Y.; Lock, L.F.; Donovan, P.J. Nucleofection mediates high-efficiency stable gene knockdown and transgene expression in human embryonic stem cells. Stem Cells 2008, 26, 1436–1443. [Google Scholar] [CrossRef]
- Landi, A.; Babiuk, L.A.; van Drunen Littel-van den Hurk, S. High transfection efficiency, gene expression, and viability of monocyte-derived human dendritic cells after nonviral gene transfer. J. Leukoc. Biol. 2007, 82, 849–860. [Google Scholar] [CrossRef]
- Recillas-Targa, F. Multiple strategies for gene transfer, expression, knockdown, and chromatin influence in mammalian cell lines and transgenic animals. Mol. Biotechnol. 2006, 34, 337–354. [Google Scholar] [CrossRef]
- Ribeiro, S.; Mairhofer, J.; Madeira, C.; Diogo, M.M.; Lobato da Silva, C.; Monteiro, G.; Grabherr, R.; Cabral, J.M. Plasmid DNA Size Does Affect Nonviral Gene Delivery Efficiency in Stem Cells. Cell. Reprogram. 2012, 14, 130–137. [Google Scholar] [CrossRef]
- Lesueur, L.L.; Mir, L.M.; André, F.M. Overcoming the Specific Toxicity of Large Plasmids Electrotransfer in Primary Cells In Vitro. Mol. Ther.-Nucleic Acids 2016, 5, e291. [Google Scholar] [CrossRef]
- Boussif, O.; Lezoualc’h, F.; Zanta, M.A.; Mergny, M.D.; Scherman, D.; Demeneix, B.; Behr, J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc. Natl. Acad. Sci. USA 1995, 92, 7297–7301. [Google Scholar] [CrossRef]
- Onawane, N.D.; Szoka, F.C.; Verkman, A.S. Chloride Accumulation and Swelling in Endosomes Enhances DNA Transfer by Polyamine-DNA Polyplexes. J. Biol. Chem. 2003, 278, 44826–44831. [Google Scholar] [CrossRef]
- Keith, M.B.A.; Farrell, P.J.; Iatrou, K.; Behie, L.A. Use of Flow Cytometry to Rapidly Optimize the Transfection of Animal Cells. BioTechniques 2000, 28, 148–154. [Google Scholar] [CrossRef]
- Shao, J.; Wang, M.; Yu, G.; Zhu, S.; Yu, Y.; Heng, B.C.; Wu, J.; Ye, H. Synthetic far-red light-mediated CRISPR-dCas9 device for inducing functional neuronal differentiation. Proc. Natl. Acad. Sci. USA 2018, 115, E6722–E6730. [Google Scholar] [CrossRef]
- Shigekawa, K.; Dower, W.J. Electroporation of eukaryotes and prokaryotes: A general approach to the introduction of macromolecules into cells. BioTechniques 1988, 6, 742–751. [Google Scholar]
- Jacobsen, F.; Mertens-Rill, J.; Beller, J.; Hirsch, T.; Daigeler, A.; Langer, S.; Lehnhardt, M.; Steinau, H.-U.; Steinstraesser, L. Nucleofection: A new method for cutaneous gene transfer? J. Biomed. Biotechnol. 2006, 2006, 26060. [Google Scholar] [CrossRef]
- Maasho, K.; Marusina, A.; Reynolds, N.M.; Coligan, J.E.; Borrego, F. Efficient gene transfer into the human natural killer cell line, NKL, using the Amaxa nucleofection system™. J. Immunol. Methods 2004, 284, 133–140. [Google Scholar] [CrossRef]
- Stewart, M.P.; Langer, R.; Jensen, K.F. Intracellular Delivery by Membrane Disruption: Mechanisms, Strategies, and Concepts. Chem. Rev. 2018, 118, 7409–7531. [Google Scholar] [CrossRef]
- Søndergaard, J.N.; Geng, K.; Sommerauer, C.; Atanasoai, I.; Yin, X.; Kutter, C. Successful delivery of large-size CRISPR/Cas9 vectors in hard-to-transfect human cells using small plasmids. Commun. Biol. 2020, 3, 319. [Google Scholar] [CrossRef]
- Saunderson, E.A.; Stepper, P.; Gomm, J.J.; Hoa, L.; Morgan, A.; Allen, M.D.; Jones, J.L.; Gribben, J.G.; Jurkowski, T.P.; Ficz, G. Hit-and-run epigenetic editing prevents senescence entry in primary breast cells from healthy donors. Nat. Commun. 2017, 8, 1450. [Google Scholar] [CrossRef] [PubMed]
- Wendt, A.; Eliasson, L. Pancreatic α-cells—The unsung heroes in islet function. Semin. Cell Dev. Biol. 2020, 103, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Srienc, F. Quantitative analysis of transient gene expression in mammalian cells using the green fluorescent protein. J. Biotechnol. 1996, 49, 137–151. [Google Scholar] [CrossRef]
- Tyumentseva, M.A.; Tyumentsev, A.I.; Akimkin, V.G. Protocol for assessment of the efficiency of CRISPR/Cas RNP delivery to different types of target cells. PLoS ONE 2021, 16, e0259812. [Google Scholar] [CrossRef] [PubMed]
- Pini, V.; Mariot, V.; Dumonceaux, J.; Counsell, J.; O’Neill, H.C.; Farmer, S.; Conti, F.; Muntoni, F. Transiently expressed CRISPR/Cas9 induces wild-type dystrophin in vitro in DMD patient myoblasts carrying duplications. Sci. Rep. 2022, 12, 3756. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zou, Y.; Zhang, L.; Tang, J.; Niedermann, G.; Firat, E.; Huang, X.; Zhu, X. Nucleofection with Plasmid DNA for CRISPR/Cas9-Mediated Inactivation of Programmed Cell Death Protein 1 in CD133-Specific CAR T Cells. Hum. Gene Ther. 2019, 30, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Yu, J. Electroporation of CRISPR-Cas9 into Malignant B Cells for Loss-of-Function Studies of Target Gene Via Knockout. In Electroporation Protocols: Microorganism, Mammalian System, and Nanodevice; Li, S., Chang, L., Teissie, J., Eds.; Springer: New York, NY, USA, 2020; pp. 85–90. [Google Scholar]
- Seki, A.; Rutz, S. Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells. J. Exp. Med. 2018, 215, 985–997. [Google Scholar] [CrossRef]
- Zoppo, M.; Okoniewski, N.; Pantelyushin, S.; vom Berg, J.; Schirmer, K. A ribonucleoprotein transfection strategy for CRISPR/Cas9-mediated gene editing and single cell cloning in rainbow trout cells. Cell Biosci. 2021, 11, 103. [Google Scholar] [CrossRef]
- Schubert, M.S.; Thommandru, B.; Woodley, J.; Turk, R.; Yan, S.; Kurgan, G.; McNeill, M.S.; Rettig, G.R. Optimized design parameters for CRISPR Cas9 and Cas12a homology-directed repair. Sci. Rep. 2021, 11, 19482. [Google Scholar] [CrossRef]
- Savell, K.E.; Bach, S.V.; Zipperly, M.E.; Revanna, J.S.; Goska, N.A.; Tuscher, J.J.; Duke, C.G.; Sultan, F.A.; Burke, J.N.; Williams, D.; et al. A Neuron-Optimized CRISPR/dCas9 Activation System for Robust and Specific Gene Regulation. Eneuro 2019, 6, ENEURO.0495-18.2019. [Google Scholar] [CrossRef]
- Carnegie, J.R.; Robert-Cooperman, C.E.; Wu, J.; Young, R.A.; Wolf, B.A.; Burkhardt, B.R. Characterization of the expression, localization, and secretion of PANDER in alpha-cells. Mol. Cell. Endocrinol. 2010, 325, 36–45. [Google Scholar] [CrossRef][Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Đorđević, M.; Paunović, V.; Jovanović Tucović, M.; Tolić, A.; Rajić, J.; Dinić, S.; Uskoković, A.; Grdović, N.; Mihailović, M.; Marković, I.; et al. Nucleofection as an Efficient Method for Alpha TC1-6 Cell Line Transfection. Appl. Sci. 2022, 12, 7938. https://doi.org/10.3390/app12157938
Đorđević M, Paunović V, Jovanović Tucović M, Tolić A, Rajić J, Dinić S, Uskoković A, Grdović N, Mihailović M, Marković I, et al. Nucleofection as an Efficient Method for Alpha TC1-6 Cell Line Transfection. Applied Sciences. 2022; 12(15):7938. https://doi.org/10.3390/app12157938
Chicago/Turabian StyleĐorđević, Marija, Verica Paunović, Maja Jovanović Tucović, Anja Tolić, Jovana Rajić, Svetlana Dinić, Aleksandra Uskoković, Nevena Grdović, Mirjana Mihailović, Ivanka Marković, and et al. 2022. "Nucleofection as an Efficient Method for Alpha TC1-6 Cell Line Transfection" Applied Sciences 12, no. 15: 7938. https://doi.org/10.3390/app12157938
APA StyleĐorđević, M., Paunović, V., Jovanović Tucović, M., Tolić, A., Rajić, J., Dinić, S., Uskoković, A., Grdović, N., Mihailović, M., Marković, I., Arambašić Jovanović, J., & Vidaković, M. (2022). Nucleofection as an Efficient Method for Alpha TC1-6 Cell Line Transfection. Applied Sciences, 12(15), 7938. https://doi.org/10.3390/app12157938