
Genetic Diversity and Population Structure of the Endangered Ulmusvillosa in Pakistan Revealed by DNA Barcode Markers

 , , ,
, , ,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
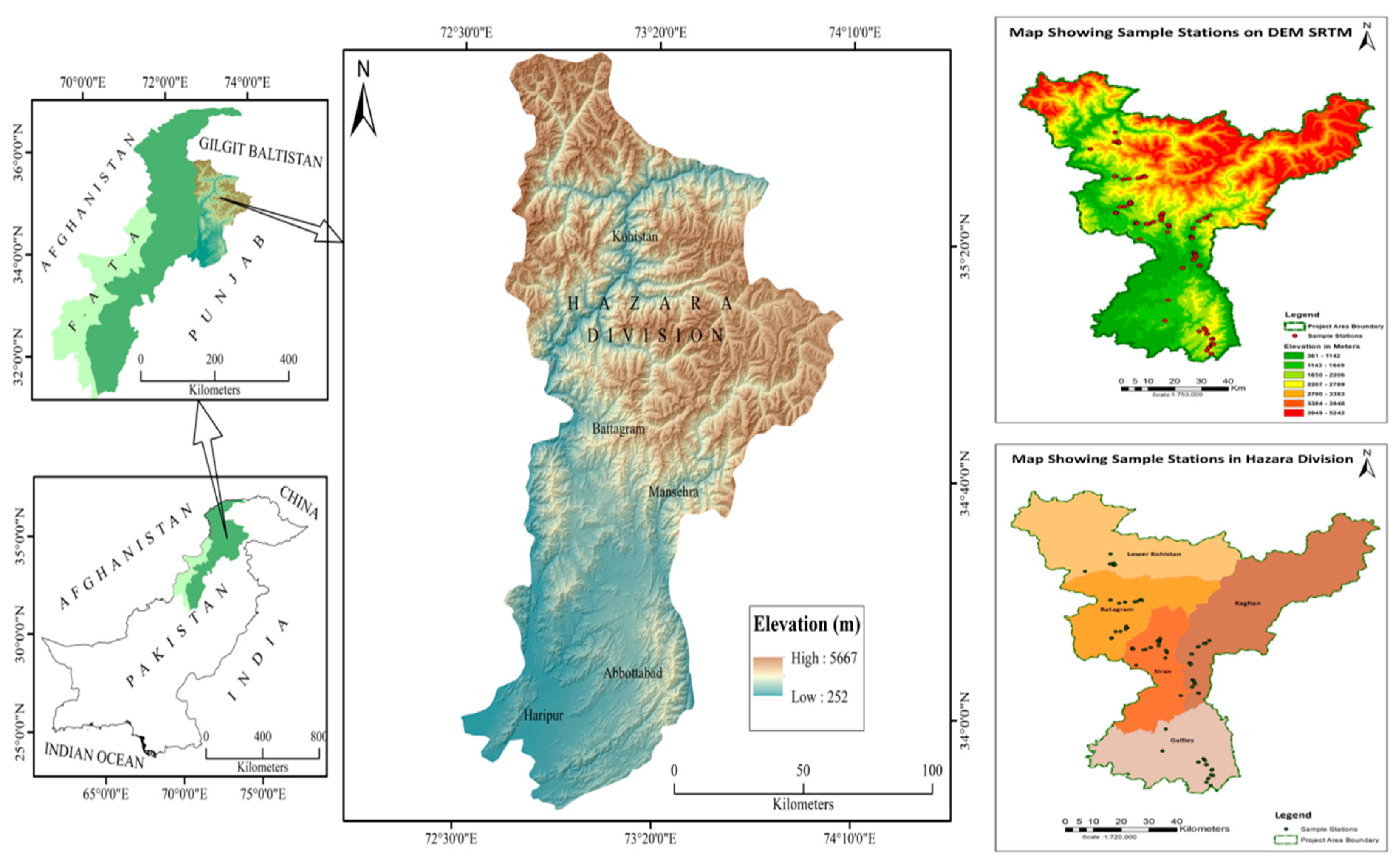
2.1. Study Area and Plant Materials
2.2. Genomic DNA Extraction and DNA Quantification
2.3. Molecular Markers Selection and PCR Analyses
2.4. Gel Purification and Confirmation of Eluted PCR Products
2.5. Data Analyses
3. Results
3.1. Molecular Markers (Barcoding Genes) Analyses
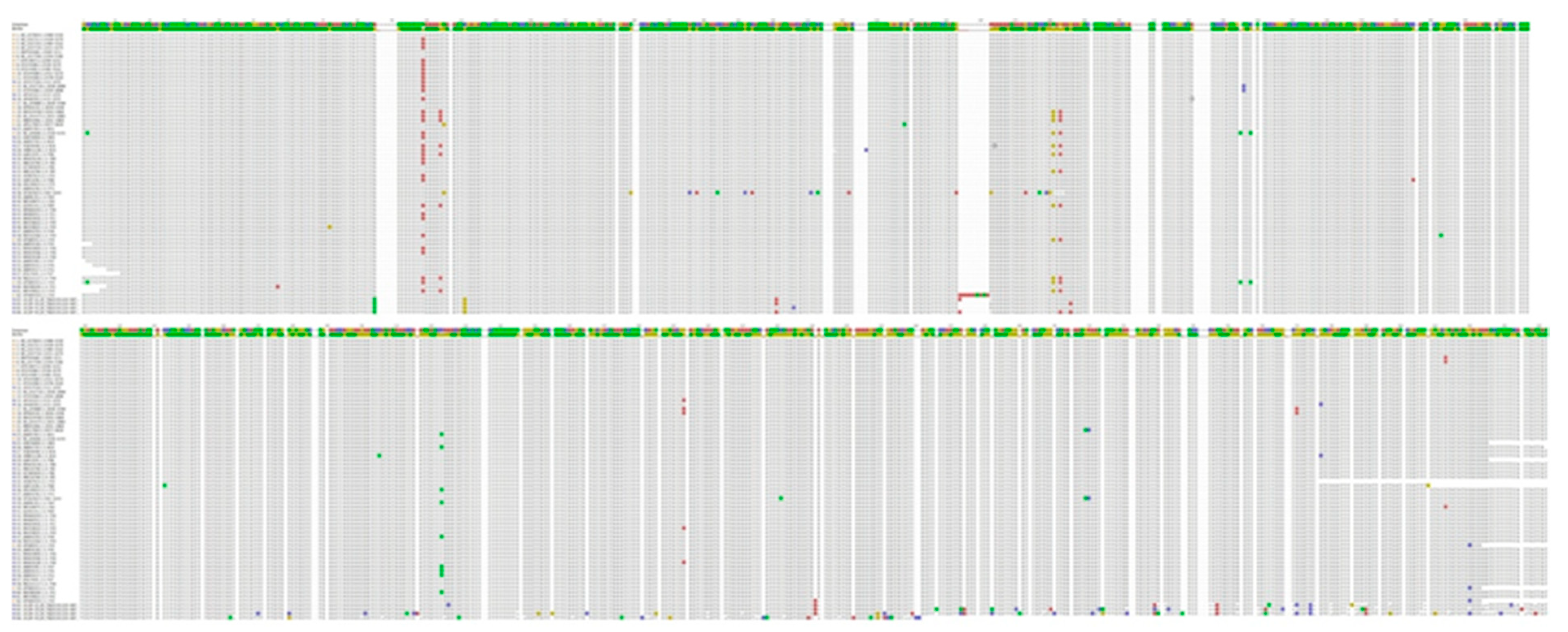
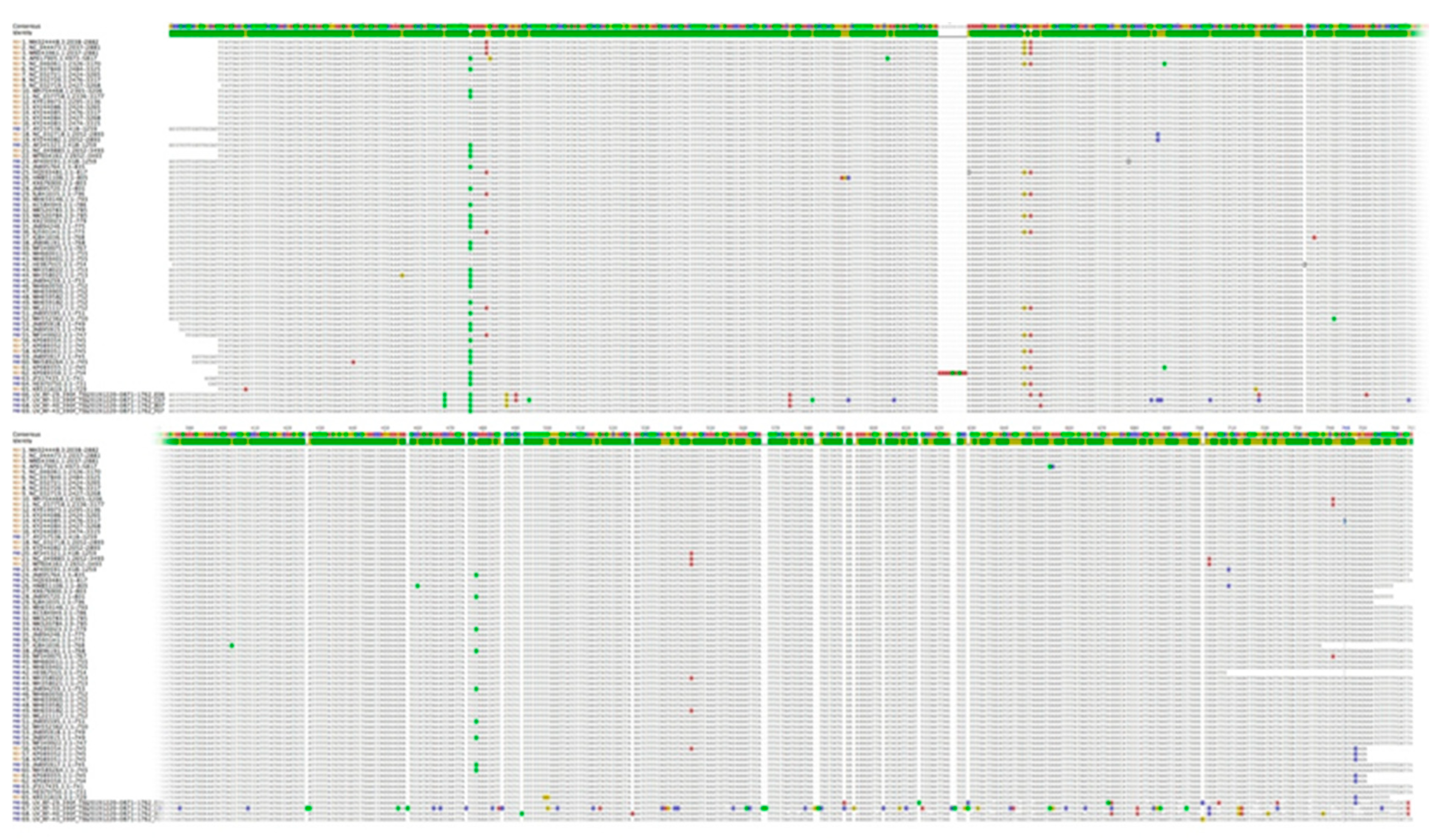
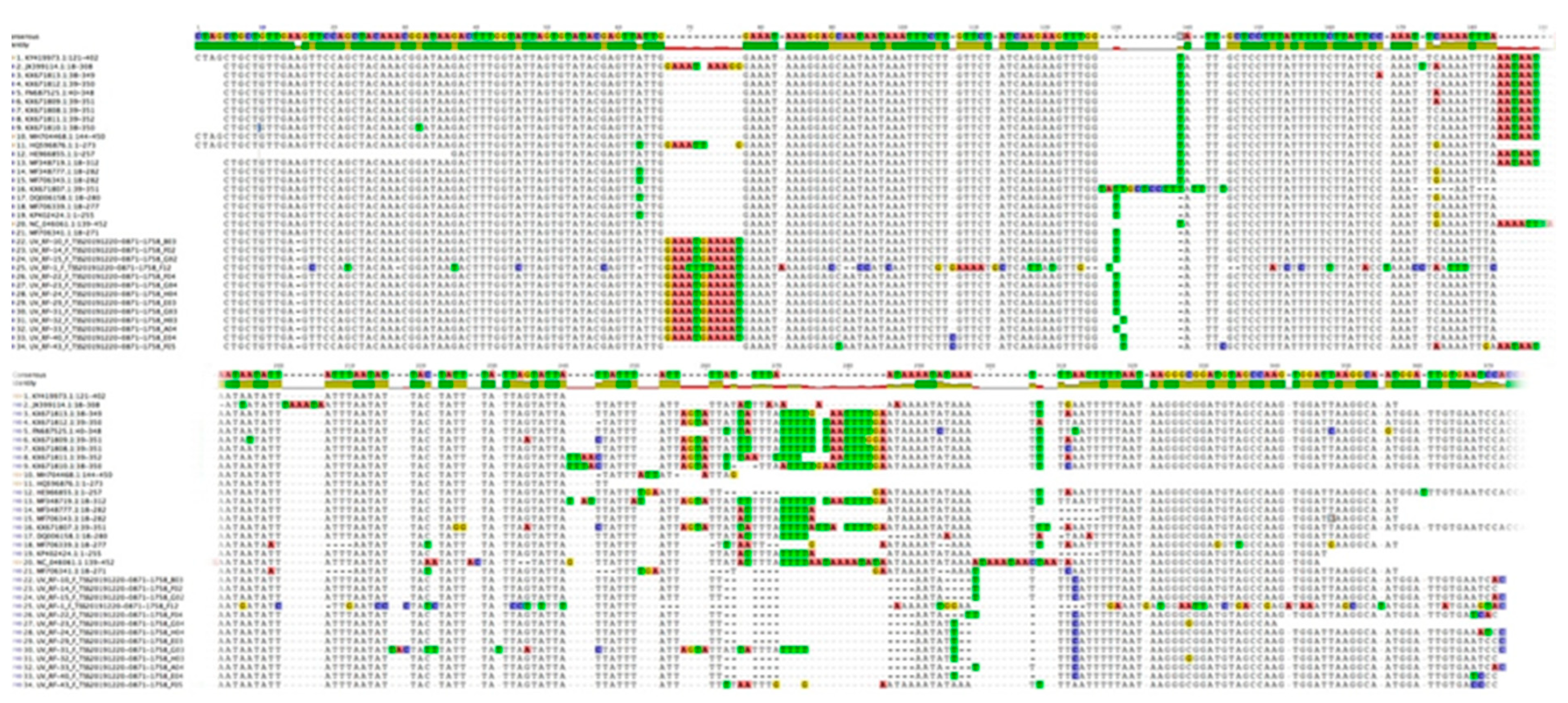
3.2. Nucleotide Sequence Variation and Phylogeny of U. villosa
3.2.1. rbcL Sequences
3.2.2. matK XF + 5R and matK 390F + 1326R Sequences
3.2.3. trnH-psbA Sequences
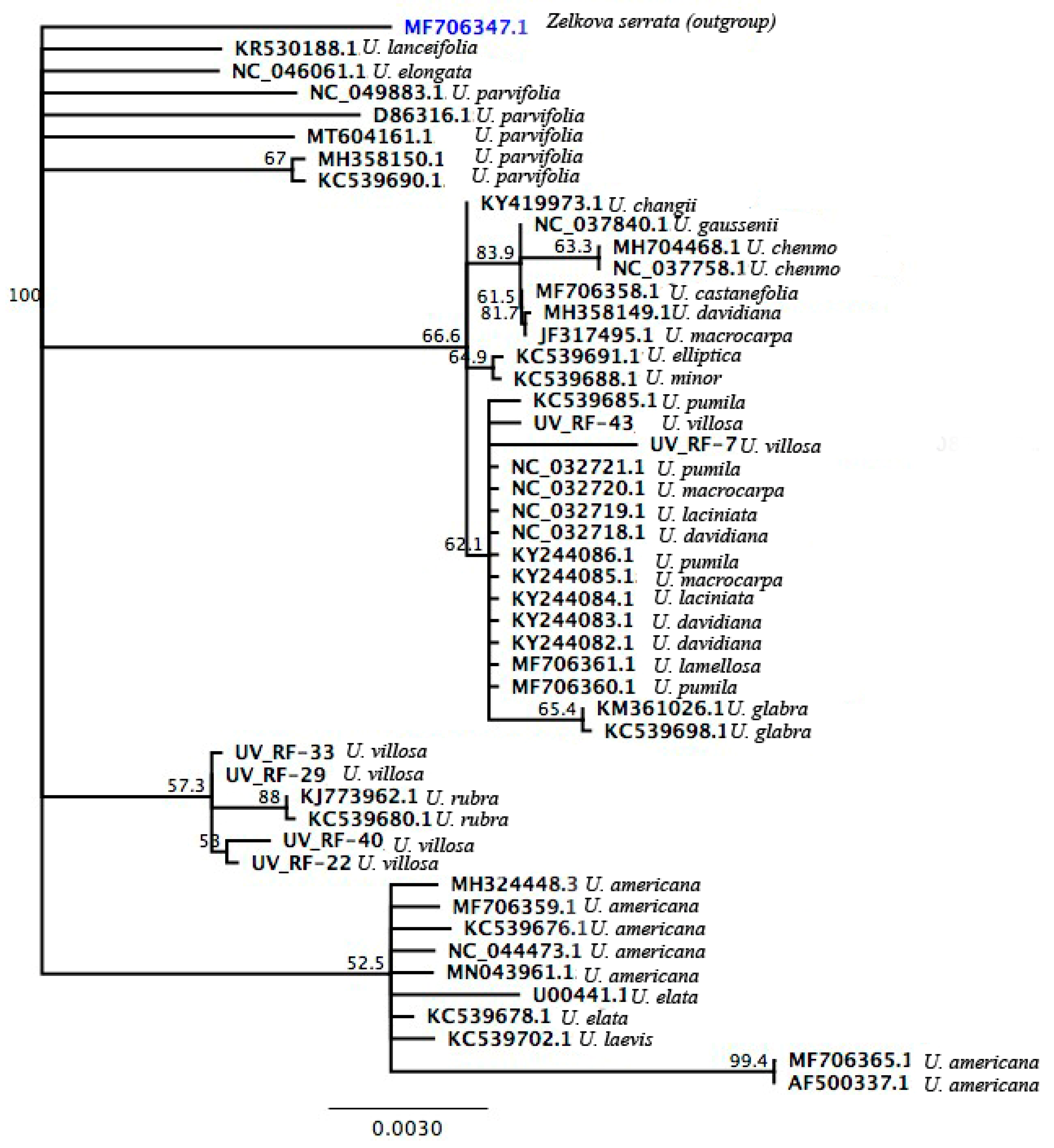
3.3. Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Richins, M.L. Negative Word-of-Mouth by Dissatisfied Consumers: A Pilot Study. J. Mark. 1983, 47, 68–78. [Google Scholar] [CrossRef]
- Khan, R.; Ali, N.; Hussain, M.; Rahman, I.U.; Majid, A.; Romman, M.; Ahmed, T.; Shah, A.H. Evaluation of the Conservation Status of Ulmus wallichiana and U. vilosa in Pakistan. Pakistan J. Bot. 2021, 53, 2127–2134. [Google Scholar] [CrossRef]
- Reisch, C.; Hartig, F. Species and Genetic Diversity Patterns Show Different Responses to Land Use Intensity in Central European Grasslands. Divers. Distrib. 2021, 27, 392–401. [Google Scholar] [CrossRef]
- Vu, D.D.; Bui, T.T.X.; Nguyen, M.T.; Vu, D.G.; Nguyen, M.D.; Bui, V.T.; Huang, X.; Zhang, Y. Genetic Diversity in Two Threatened Species in Vietnam: Taxus chinensis and Taxus wallichiana. J. For. Res. 2017, 28, 265–272. [Google Scholar] [CrossRef]
- Vu, D.D.; Bui, T.T.X.; Nguyen, M.D.; Shah, S.N.M.; Vu, D.G.; Zhang, Y.; Nguyen, M.T.; Huang, X.H. Genetic Diversity and Conservation of Two Threatened Dipterocarps (Dipterocarpaceae) in Southeast Vietnam. J. For. Res. 2019, 30, 1823–1831. [Google Scholar] [CrossRef]
- Nguyen, T.M.; Vu, D.D.; Dang, H.P.; Bui, X.T.T.; Nguyen, H.P.L.; Nguyen, D.M. Population Genetic Structure and Demographic History of the Dipterocarp Species Anisoptera Costata Korth Revealed by Microsatellite Analysis. Planta 2021, 253, 66. [Google Scholar] [CrossRef] [PubMed]
- Frankham, R.; Ballou, J.D.; Briscoe, D.A.; McInnes, K.H. Introduction to Conservation Genetics; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- Petit, R.J.; El Mousadik, A.; Pons, O. Identifying Populations for Conservation on the Basis of Genetic Markers. Conserv. Biol. 1998, 12, 844–855. [Google Scholar] [CrossRef]
- Wiegrefe, S.J.; Sytsma, K.J.; Guries, R.P. Phylogeny of Elms (Ulmus, Ulmaceae): Molecular Evidence for a Sectional Classification. Syst. Bot. 1994, 19, 590–612. [Google Scholar] [CrossRef]
- Machon, N.; Lefranc, M.; Bilger, I.; Mazer, S.J.; Sarr, A. Allozyme Variation in Ulmus Species from France: Analysis of Differentiation. Heredity 1997, 78, 12–20. [Google Scholar] [CrossRef]
- Cogolludo-Agustín, M.Á.; Agúndez, D.; Gil, L. Identification of Native and Hybrid Elms in Spain Using Isozyme Gene Markers. Heredity 2000, 85, 157–166. [Google Scholar] [CrossRef][Green Version]
- Goodall-Copestake, W.P.; Hollingsworth, M.L.; Hollingsworth, P.M.; Jenkins, G.I.; Collin, E. Molecular Markers and Ex Situ Conservation of the European Elms (Ulmus Spp.). Biol. Conserv. 2005, 122, 537–546. [Google Scholar] [CrossRef]
- Pooler, M.R.; Townsend, A.M. DNA Fingerprinting of Clones and Hybrids of American Elm and Other Elm Species with AFLP Markers. J. Environ. Hortic. 2005, 23, 113–117. [Google Scholar] [CrossRef]
- Collada, C.; Fuentes-Utrilla, P.; Gil, L.; Cervera, M.T. Characterization of Microsatellite Loci in Ulmus minor Miller and Cross-Amplification in U. Glabra Hudson and U. Laevis Pall. Mol. Ecol. Notes 2004, 4, 731–732. [Google Scholar] [CrossRef]
- Zalapa, J.E.; Brunet, J.; Guries, R.P. Isolation and Characterization of Microsatellite Markers for Red Elm (Ulmus rubra Muhl.) and Cross-Species Amplification with Siberian Elm (Ulmus pumila L.). Mol. Ecol. Resour. 2008, 8, 109–112. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; deWaard, J.R. Biological Identifications through DNA Barcodes. Proceedings. Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef]
- Kress, W.J.; Erickson, D.L. A Two-Locus Global DNA Barcode for Land Plants: The Coding RbcL Gene Complements the Non-Coding TrnH-PsbA Spacer Region. PLoS ONE 2007, 2, e508. [Google Scholar] [CrossRef]
- De Moraes Russo, C.A.; Selvatti, A.P. Bootstrap and Rogue Identification Tests for Phylogenetic Analyses. Mol. Biol. Evol. 2018, 35, 2327–2333. [Google Scholar] [CrossRef]
- Kress, W.J.; Erickson, D.L. DNA Barcodes: Genes, Genomics, and Bioinformatics. Proc. Natl. Acad. Sci. USA. 2008, 105, 2761–2762. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Tuimala, J. A Primer to Phylogenetic Analysis Using the PHYLIP Package; Center for Scientific Computing Ltd: Espoo, Finland, 2004. [Google Scholar]
- Kress, W.J. Plant DNA Barcodes: Applications Today and in the Future. J. Syst. Evol. 2017, 55, 291–307. [Google Scholar] [CrossRef]
- Nithaniyal, S.; Parani, M. Evaluation of Chloroplast and Nuclear DNA Barcodes for Species Identification in Terminalia L. Biochem. Syst. Ecol. 2016, 68, 223–229. [Google Scholar] [CrossRef]
- Li, M.; Chen, Q.; Zhang, L.; Guo, P.; Wang, Y. The Complete Chloroplast Genome Sequence of Ulmus parvifolia (Ulmaceae). Mitochondrial DNA. Part B Resour. 2020, 5, 2957–2958. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Jiao, L.; Guo, J.; Wiedenhoeft, A.C.; He, T.; Jiang, X.; Yin, Y. DNA Barcoding of Vouchered Xylarium Wood Specimens of Nine Endangered Dalbergia Species. Planta 2017, 246, 1165–1176. [Google Scholar] [CrossRef] [PubMed]
- Lahaye, R.; van der Bank, M.; Bogarin, D.; Warner, J.; Pupulin, F.; Gigot, G.; Maurin, O.; Duthoit, S.; Barraclough, T.G.; Savolainen, V. DNA Barcoding the Floras of Biodiversity Hotspots. Proc. Natl. Acad. Sci. USA 2008, 105, 2923–2928. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, R.; Geng, M.; Qin, Y.; Liu, H.; Li, M. The Complete Chloroplast Genome of Zelkova Serrata and Its Phylogenetic Position within Ulmaceae. Mitochondrial DNA. Part B Resour. 2020, 5, 2182–2183. [Google Scholar] [CrossRef]
- Shuvaev, S.A.; Başerdem, B.; Zador, A.M.; Koulakov, A.A. Network Cloning Using DNA Barcodes. Proc. Natl. Acad. Sci. USA 2019, 116, 9610–9615. [Google Scholar] [CrossRef]
- Healey, A.; Furtado, A.; Cooper, T.; Henry, R.J. Protocol: A Simple Method for Extracting next-Generation Sequencing Quality Genomic DNA from Recalcitrant Plant Species. Plant Methods 2014, 10, 21. [Google Scholar] [CrossRef]
- Cuénoud, P.; Savolainen, V.; Chatrou, L.W.; Powell, M.; Grayer, R.J.; Chase, M.W. Molecular Phylogenetics of Caryophyllales Based on Nuclear 18S RDNA and Plastid RbcL, AtpB, and MatK DNA Sequences. Am. J. Bot. 2002, 89, 132–144. [Google Scholar] [CrossRef]
- Chase, M.W.; Cowan, R.S.; Hollingsworth, P.M.; Van Den Berg, C.; Madriñán, S.; Petersen, G.; Seberg, O.; Jørgsensen, T.; Cameron, K.M.; Carine, M.; et al. A Proposal for a Standardised Protocol to Barcode All Land Plants. Taxon 2007, 56, 295–299. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. | Barcode/Marker Name | Sequence (5-3′) | Optimum Temperature °C | Amplified Product Size |
|---|---|---|---|---|
| 1 | rbcL *a | 1F: ATGTCACCACAAACAGAAAC 724R: TCGCATGTACCTGCAGTAGC | 52 | 726 bp |
| 2 | matK *a | XF: TAATTTACGATCAATTCATTC 5R: GTTCTAGCACAAGAAAGTCG | 52 | 802 bp |
| 3 | matK *a | 390F: CGATCTATTCATTCAATATTTC 1326R: TCTAGCACACGAAAGTCGAAGT | 49.5 | 966 bp |
| 4 | trnH-psbA *b | F: GTTATGCATGAACGTAATGCTC R: CGCGCATGGTGGATTCACAATCC | 55 | 651 bp |
| Region/Sequence Name | Total Sequences Included | % Pairwise Identity | % Identical Sites | Max Length (bp) | Min Sequence Length (bp) |
|---|---|---|---|---|---|
| matK 390F + 1326R | 69 | 98.9 | 80.3 | 739 | 680 |
| rbcL | 49 | 99.2 | 95.8 | 686 | 630 |
| trnH-psbA | 34 | 85.1 | 43.4 | 310 | 195 |
| matK XF + 5R | 68 | 99.0 | 81.5 | 784 | 574 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, R.U.; Ali, N.; Rahman, S.U.; Rahman, I.U.; Hashem, A.; Almutairi, K.F.; Fathi Abd_Allah, E.; Harsonowati, W.; Khan, M.A.; Rahim, F.; et al. Genetic Diversity and Population Structure of the Endangered Ulmusvillosa in Pakistan Revealed by DNA Barcode Markers. Appl. Sci. 2022, 12, 9293. https://doi.org/10.3390/app12189293
Khan RU, Ali N, Rahman SU, Rahman IU, Hashem A, Almutairi KF, Fathi Abd_Allah E, Harsonowati W, Khan MA, Rahim F, et al. Genetic Diversity and Population Structure of the Endangered Ulmusvillosa in Pakistan Revealed by DNA Barcode Markers. Applied Sciences. 2022; 12(18):9293. https://doi.org/10.3390/app12189293
Chicago/Turabian StyleKhan, Rafi Ullah, Niaz Ali, Siddiq Ur Rahman, Inayat Ur Rahman, Abeer Hashem, Khalid F. Almutairi, Elsayed Fathi Abd_Allah, Wiwiek Harsonowati, Muazzam Ali Khan, Fazli Rahim, and et al. 2022. "Genetic Diversity and Population Structure of the Endangered Ulmusvillosa in Pakistan Revealed by DNA Barcode Markers" Applied Sciences 12, no. 18: 9293. https://doi.org/10.3390/app12189293
APA StyleKhan, R. U., Ali, N., Rahman, S. U., Rahman, I. U., Hashem, A., Almutairi, K. F., Fathi Abd_Allah, E., Harsonowati, W., Khan, M. A., Rahim, F., Khan, F. U., & Haq, N. u. (2022). Genetic Diversity and Population Structure of the Endangered Ulmusvillosa in Pakistan Revealed by DNA Barcode Markers. Applied Sciences, 12(18), 9293. https://doi.org/10.3390/app12189293