
Monitoring Molecular Properties of a Fluorescence Light-Up Aptamer Using Fluorescence Cross-Correlation Spectroscopy



Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of the RNA
2.2. Sample Preparation
2.3. FCS and FCCS Measurement
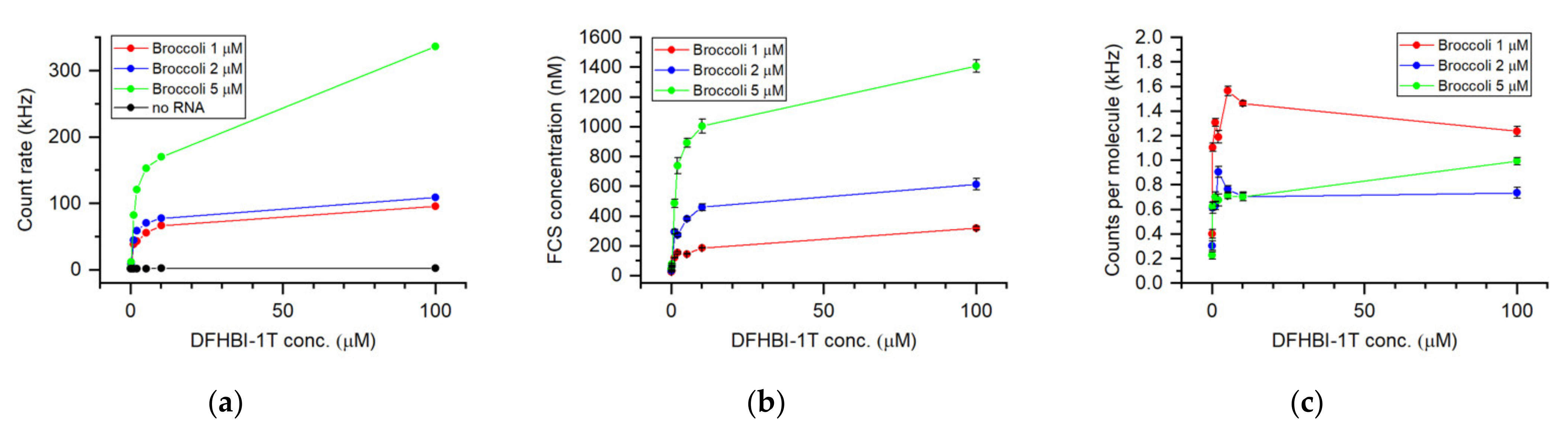
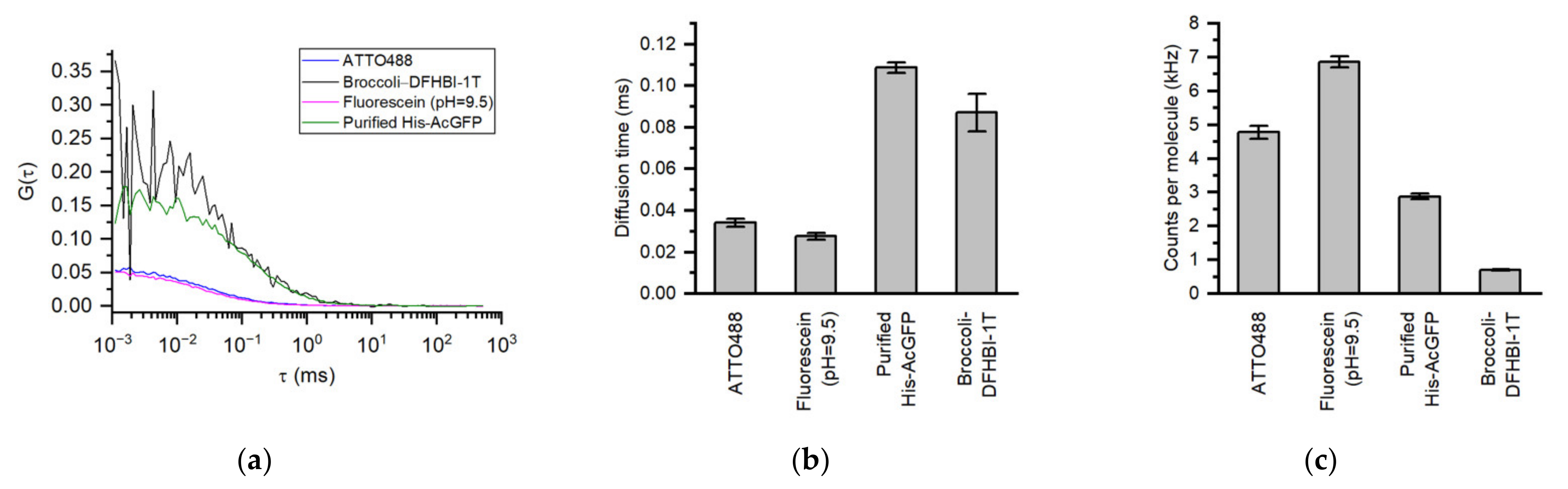
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Filonov, G.S.; Moon, J.D.; Svensen, N.; Jaffrey, S.R. Broccoli: Rapid selection of an RNA mimic of green fluorescent protein by fluorescence-based selection and directed evolution. J. Am. Chem. Soc. 2014, 136, 16299–16308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truong, L.; Ferré-D’Amaré, A.R. From fluorescent proteins to fluorogenic RNAs: Tools for imaging cellular macromolecules. Protein Sci. 2019, 28, 1374–1386. [Google Scholar] [CrossRef] [PubMed]
- Neubacher, S.; Hennig, S. RNA Structure and Cellular Applications of Fluorescent Light-Up Aptamers. Angew. Chem. Int. Ed. Engl. 2019, 58, 1266–1279. [Google Scholar] [CrossRef] [PubMed]
- Elson, E.L.; Magde, D. Fluorescence correlation spectroscopy. I. Conceptual basis and theory. Biopolymers 1974, 13, 1–27. [Google Scholar] [CrossRef]
- Magde, D.; Elson, E.L.; Webb, W.W. Fluorescence correlation spectroscopy. II. An experimental realization. Biopolymers 1974, 13, 29–61. [Google Scholar] [CrossRef]
- Rigler, R.; Mets, U.; Widengren, J.; Kask, P. Fluorescence Correlation Spectroscopy with High Count Rate and Low-Background—Analysis of Translational Diffusion. Eur. Biophys. J. Biophys. Lett. 1993, 22, 169–175. [Google Scholar] [CrossRef]
- Mateu-Regué, À.; Christiansen, J.; Bagger, F.O.; Hellriegel, C.; Nielsen, F.C. Unveiling mRNP composition by fluorescence correlation and cross-correlation spectroscopy using cell lysates. Nucleic Acids Res. 2021, 49, e119. [Google Scholar] [CrossRef]
- Imamura, H.; Sasaki, A.; Honda, S. Fate of a Stressed Therapeutic Antibody Tracked by Fluorescence Correlation Spectroscopy: Folded Monomers Survive Aggregation. J. Phys. Chem. B 2017, 121, 8085–8093. [Google Scholar] [CrossRef]
- Sasaki, A.; Yamamoto, J.; Kinjo, M.; Noda, N. Absolute Quantification of RNA Molecules Using Fluorescence Correlation Spectroscopy with Certified Reference Materials. Anal. Chem. 2018, 90, 10865–10871. [Google Scholar] [CrossRef] [Green Version]
- Kinjo, M.; Rigler, R. Ultrasensitive Hybridization Analysis Using Fluorescence Correlation Spectroscopy. Nucleic Acids Res. 1995, 23, 1795–1799. [Google Scholar] [CrossRef]
- Rigler, R.; Földes-Papp, Z.; Meyer-Almes, F.-J.; Sammet, C.; Völcker, M.; Schnetz, A. Fluorescence cross-correlation: A new concept for polymerase chain reaction. J. Biotechnol. 1998, 63, 97–109. [Google Scholar] [CrossRef]
- Bacia, K.; Kim, S.A.; Schwille, P. Fluorescence cross-correlation spectroscopy in living cells. Nat. Methods 2006, 3, 83–89. [Google Scholar] [CrossRef]
- Tiwari, M.; Mikuni, S.; Muto, H.; Kinjo, M. Determination of dissociation constant of the NFκB p50/p65 heterodimer using fluorescence cross-correlation spectroscopy in the living cell. Biochem. Biophys. Res. Commun. 2013, 436, 430–435. [Google Scholar] [CrossRef]
- Sadaie, W.; Harada, Y.; Matsuda, M.; Aoki, K. Quantitative in vivo fluorescence cross-correlation analyses highlight the importance of competitive effects in the regulation of protein-protein interactions. Mol. Cell Biol. 2014, 34, 3272–3290. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, A.; Kinjo, M. Monitoring intracellular degradation of exogenous DNA using diffusion properties. J. Control Release 2010, 143, 104–111. [Google Scholar] [CrossRef]
- Dertinger, T.; Pacheco, V.; von der Hocht, I.; Hartmann, R.; Gregor, I.; Enderlein, J. Two-focus fluorescence correlation spectroscopy: A new tool for accurate and absolute diffusion measurements. ChemPhysChem 2007, 8, 433–443. [Google Scholar] [CrossRef]
- Culbertson, C.T.; Jacobson, S.C.; Michael Ramsey, J. Diffusion coefficient measurements in microfluidic devices. Talanta 2002, 56, 365–373. [Google Scholar] [CrossRef]
- Wang, P.; Querard, J.; Maurin, S.; Nath, S.; Le Saux, T.; Gautier, A.; Jullien, L. Photochemical properties of Spinach and its use in selective imaging. Chem. Sci. 2013, 4, 2865–2873. [Google Scholar] [CrossRef]
- Han, K.Y.; Leslie, B.J.; Fei, J.; Zhang, J.; Ha, T. Understanding the photophysics of the spinach-DFHBI RNA aptamer-fluorogen complex to improve live-cell RNA imaging. J. Am. Chem. Soc. 2013, 135, 19033–19038. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wu, J.; Jaffrey, S.R. Engineering Fluorophore Recycling in a Fluorogenic RNA Aptamer. Angew. Chem. Int. Ed. Engl. 2021, 60, 24153–24161. [Google Scholar] [CrossRef]
- Dolgosheina, E.V.; Jeng, S.C.; Panchapakesan, S.S.; Cojocaru, R.; Chen, P.S.; Wilson, P.D.; Hawkins, N.; Wiggins, P.A.; Unrau, P.J. RNA mango aptamer-fluorophore: A bright, high-affinity complex for RNA labeling and tracking. ACS Chem. Biol. 2014, 9, 2412–2420. [Google Scholar] [CrossRef]
- Autour, A.; Jeng, S.Y.C.; Cawte, A.D.; Abdolahzadeh, A.; Galli, A.; Panchapakesan, S.S.S.; Rueda, D.; Ryckelynck, M.; Unrau, P.J. Fluorogenic RNA Mango aptamers for imaging small non-coding RNAs in mammalian cells. Nat. Commun. 2018, 9, 656. [Google Scholar] [CrossRef]
- Song, W.; Filonov, G.S.; Kim, H.; Hirsch, M.; Li, X.; Moon, J.D.; Jaffrey, S.R. Imaging RNA polymerase III transcription using a photostable RNA-fluorophore complex. Nat. Chem. Biol. 2017, 13, 1187–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirth, R.; Gao, P.; Nienhaus, G.U.; Sunbul, M.; Jäschke, A. SiRA: A Silicon Rhodamine-Binding Aptamer for Live-Cell Super-Resolution RNA Imaging. J. Am. Chem. Soc. 2019, 141, 7562–7571. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, D.; Su, N.; Bao, B.; Xie, X.; Zuo, F.; Yang, L.; Wang, H.; Jiang, L.; Lin, Q.; et al. Visualizing RNA dynamics in live cells with bright and stable fluorescent RNAs. Nat. Biotechnol. 2019, 37, 1287–1293. [Google Scholar] [CrossRef] [PubMed]
- Sunbul, M.; Lackner, J.; Martin, A.; Englert, D.; Hacene, B.; Grün, F.; Nienhaus, K.; Nienhaus, G.U.; Jäschke, A. Super-resolution RNA imaging using a rhodamine-binding aptamer with fast exchange kinetics. Nat. Biotechnol. 2021, 39, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Furuhata, Y.; Kobayashi, M.; Maruyama, R.; Sato, Y.; Makino, K.; Michiue, T.; Yui, H.; Nishizawa, S.; Yoshimoto, K. Programmable RNA detection with a fluorescent RNA aptamer using optimized three-way junction formation. RNA 2019, 25, 590–599. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RCA Gc(0)/Gr(0) | RCA Gc(0)/Gg(0) | Cgreen (nM) | Cred (nM) | Ccomplex (nM) | Apparent Kd from FCCS (nM) | Estimated Ccomplex Assuming Kd = 360 nM (nM) | Folding Efficiency (Ccomplex/Estimated Ccomplex) | |
|---|---|---|---|---|---|---|---|---|
| Broccoli-ATTO647N (1 µM) /DFHBI-1T (10 µM) | 0.987 ± 0.041 | 0.198 ± 0.011 | 75.3 ± 0.0 | 260.5 ± 5.1 | 60.6 ± 2.5 | 42,961 * | 251.3 | 0.241 |
| Broccoli-ATTO647N (1 µM) /DFHBI-1T (1 µM) | 0.923 ± 0.053 | 0.137 ± 0.009 | 50.2 ± 2.7 | 245.0 ± 8.3 | 37.9 ± 3.6 | 6472 * | 170.8 | 0.222 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furuhata, Y.; Sasaki, A. Monitoring Molecular Properties of a Fluorescence Light-Up Aptamer Using Fluorescence Cross-Correlation Spectroscopy. Appl. Sci. 2022, 12, 2002. https://doi.org/10.3390/app12042002
Furuhata Y, Sasaki A. Monitoring Molecular Properties of a Fluorescence Light-Up Aptamer Using Fluorescence Cross-Correlation Spectroscopy. Applied Sciences. 2022; 12(4):2002. https://doi.org/10.3390/app12042002
Chicago/Turabian StyleFuruhata, Yuichi, and Akira Sasaki. 2022. "Monitoring Molecular Properties of a Fluorescence Light-Up Aptamer Using Fluorescence Cross-Correlation Spectroscopy" Applied Sciences 12, no. 4: 2002. https://doi.org/10.3390/app12042002
APA StyleFuruhata, Y., & Sasaki, A. (2022). Monitoring Molecular Properties of a Fluorescence Light-Up Aptamer Using Fluorescence Cross-Correlation Spectroscopy. Applied Sciences, 12(4), 2002. https://doi.org/10.3390/app12042002