1. Introduction
The cross-linking polymerization of multifunctional aromatic monomers initiated by exposure to high-energy radiation continues to be explored as a promising alternative to thermal curing for the production of high-performance composite materials [
1,
2,
3,
4]. High-energy radiation processing offers technical and economic advantages over thermosetting technology by allowing for fast and out-of-autoclave curing operation applicable to large and complex structures. It induces lower energy consumption and reduces development costs. Curing times are shorter, and resin formulations have longer shelf lives. The absence of highly volatile monomers reduces industrial and sanitary risks in the process. Radiation-induced curing is based on free radical and cationic chemistries commonly used for coating applications but not for the fabrication of massive composite parts, aside from some specific applications such as dental restorative materials [
5]. As the typical thickness for structural composites ranges from few millimeters to centimeters and requires the use of shaping tools such as metal molds, the penetration of radiation, energy deposition and thermal effects are completely different from the standard conditions for 5 to 100 μm thick strongly pigmented ink and paints cured by exposure to ultraviolet (UV) or electron beam (EB) radiation [
6]. Moreover, the requirements in terms of thermophysical and mechanical properties as well as resistance to ageing under normal operation conditions are extremely different. This results in the need for deep knowledge on the relationships between resin composition, processing conditions and resulting properties for the cured matrix and at the interfaces between the matrix and fillers or fibers [
7].
Epoxy resins form a large class of multifunctional monomers and prepolymers possessing reactive 1,2-epoxide functions (oxiranes). They are most commonly produced by the reaction under basic conditions of epichlorohydrin with various types of phenols, such as bis-phenol-A, leading to diglycidyl ether of bis-phenol A, DGEBA [
8,
9]. Another approach consists of the epoxidation of unsaturated hydrocarbon segments yielding various types of aliphatic and alicyclic epoxy monomers, such as epoxidized soya or linseed oils and cyclohexene-based esters [
10]. These two sub-classes of epoxy resins are converted into solid networks by a variety of chemical mechanisms, yet with their specific reactivity and time–temperature conversion dependence due to blend composition and vitrification behavior.
Monomers and prepolymers fitted with two or more epoxy functions indeed form covalent networks either by step-growth polymerization or by chain-growth ring opening polymerization through cationic or anionic ring-opening reactions, the latter being essentially used for the linear polymerization of epoxy monomers having a simple structure (
Scheme 1). The two modes of polymerization by chain and by stepwise growth proceed through drastically different mechanisms.
In ionic chain polymerization, there is no spontaneous chain termination such as the recombination of free radical species. Additionally, cationic polymerization is a pseudo-living process, with active centers which are accumulated in the medium as the initiation takes place (no steady state concentration). Those active centers survive and can continue propagation as long as monomer functions are still present, provided that mobility of the reactive species (cationic centers and monomers) is not restricted [
11,
12]. Once the production of new initiating centers has ceased, cationic polymerization continues by the so-called dark cure phenomenon [
13], until the active centers are trapped in the glassy network or quenched by some inhibitor entering the material. This pseudo-living behavior enables further thermal activation giving rise to some beneficial post-cure effect [
14,
15]. As a consequence of the chain-growth mechanisms of epoxies initiated by Lewis or Brønsted acids, it is possible to control the polymerization process at the different stages: from the initiation to the propagation either by the active chain end mechanism (ACE), where the neutral monomer adds to the cyclic onium chain end by a nucleophilic attack, or by the activated monomer (AM), where a stable hydroxylic terminal group adds to a protonated monomer, with the possibility of forcing transfer reactions which may favorably affect the architecture and the cross-link density of the formed network (
Scheme 2).
Most of the industrial solutions are currently based on robust systems that polymerize upon exposure to an energetic stimulus by activating latent onium salt initiators that are sensitive to high temperatures or to different types of radiation (UV-visible, accelerated electrons, X-rays and γ-rays) [
15,
16,
17].
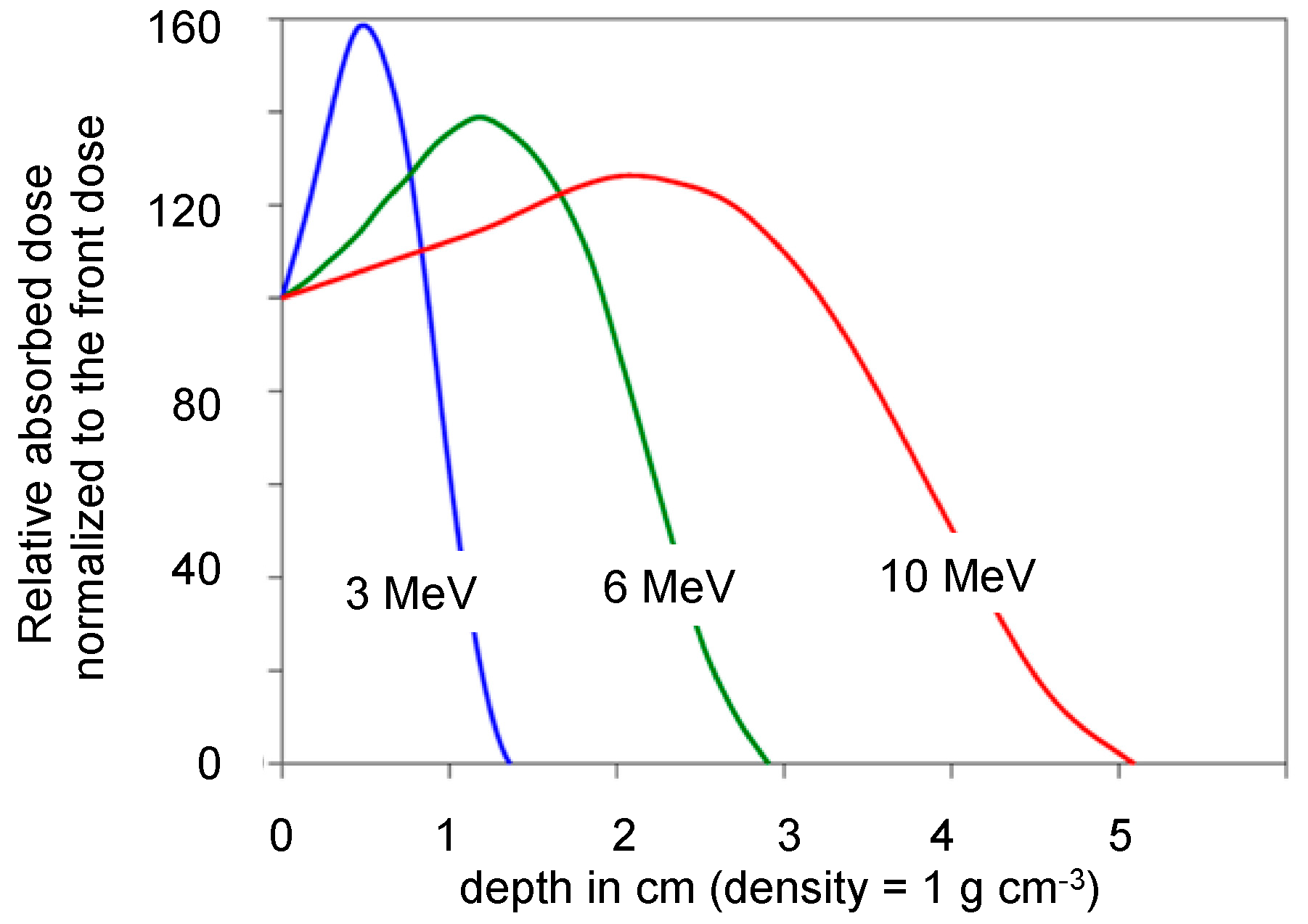
The cationic chain polymerization process which was selected for our current work will be preferably triggered by exposure to high-energy irradiation which efficiently induces the polymerization of epoxy resins. The choice for an appropriate acceleration voltage is primarily guided by the need for sufficient penetration of the electron beam into the uncured material shaped by the mold and associated tooling. The plots of
Figure 1 represent the simulated deposition of energy in a material of unit density as a function of the EB energy. This shows that composite materials of thicknesses or cross-sections up to some centimeters can be treated with a rather uniform energy deposition, provided that the mold itself does not abate dramatically the incident beam. This is actually a strong advantage of this technology operated initially at room temperature (RT) and which does not to require strong and complex tools. Moreover, lower energy EB (few hundreds eV) can, however, be used for laboratory test experiments, particularly for filler-free resin samples or simple pre-impregnated fabrics which do not require metal casting tools.
Other processing parameters include the power of the accelerator and the features of the beam (scan rate and dimensions, pulse frequency and duration), which all affect the instant and time-average dose rate [
18].
Figure 1.
Simulated depth–dose distribution (expressed as percent relative of the front dose absorbed at the entrance face of an object exposed of unit density exposed to EB radiation of energy 3, 6 and 10 MeV (adapted from Ref. [
19]).
Figure 1.
Simulated depth–dose distribution (expressed as percent relative of the front dose absorbed at the entrance face of an object exposed of unit density exposed to EB radiation of energy 3, 6 and 10 MeV (adapted from Ref. [
19]).
The interaction between matter and the high-energy photons or particles is essentially a non-selective process producing randomly excited states, radical cations and thermalized electrons as the primary species. In organic materials, the primary species are essentially converted into free radicals. Long-lived cationic chain reactions due to carbenium or oxonium intermediates generated and evidenced at low temperature in ultra-pure epoxy monomers free from any initiator are unlikely. The high reactivity of cationic centers renders transfer and deactivation reactions very fast unless special conditions (purity of involved chemicals, low temperature) are maintained throughout the process. There are still debates on the precise mechanism of cationic initiation under EB irradiation; however, when irradiation is applied to epoxies formulated with onium salts, a reasonable description of the initiation is based on an electron transfer from the in situ generated free radicals (with appropriate reduction potential) or from thermalized electrons that reduce onium salts efficiently [
18]. These mechanisms were confirmed by pulse radiolysis experiments on DGEBA in the presence of an iodonium salt [
17,
20].
For this reason, onium salts, in particular diaryliodonium salts, which exhibit favorable redox properties in the presence of organic free radicals, are introduced in the epoxy formulations processed by high-energy radiation. As similar redox reactivity of iodonium and sulfonium redox agents plays a central role under alternative modes of activation, the thioxanthone-photosensitized reduction under exposure to UV-visible photoactivation, as well as the thermal decomposition of peroxides forming reductive free radicals, can be used for the purpose of comparison or for facilitating the preparation of test samples. This is summarized in
Scheme 3 which illustrates the possible pathways to the cationic initiation of epoxide polymerization. It should be kept in mind that behind the mechanistic similarities in the chemical initiation mechanism and polymerization schemes, the actual temporal, spatial and physical conditions of these modes of activation are quite different, with significant differences in their applicability and in terms of the resulting material.
Epoxy resins cured by conventional step-growth mechanisms can be formulated and processed to yield a set of unique chemical and physical properties, including excellent chemical resistance, heat resistance, limited shrinkage on curing, high mechanical properties (strength and tenacity), low dielectric dissipation and good adhesion to polar substrates and filler [
9]. The resulting networks find applications as adhesives and matrices in composites for aerospace, automotive, electronics, and packaging sectors. Serious limitations are nevertheless associated with the use of epoxy-based matrices cured by chain reaction. Network brittleness drives the need for new systems either based on revisited monomer chemistry or based on blending conventional monomers with reactive polymers [
21]. This article covers the basic aspects of radiation curing by cationic polymerization of aromatic epoxies, and provides a status report on recent investigations implemented to improve composite properties and to enhance the overall control on the curing process.
We present here an overview of representative results obtained by curing radiation-curable formulations based on the aromatic epoxy resin DGEBA with an n value close to zero, whereas in practice formulations containing DGEBA oligomers, epoxynovolak (EPN) and solid epoxy prepolymer DGEBA-Novolak-type can be formulated to adjust the initial viscosity, reactivity and modulus of the cured materials (
Scheme 4a). In situ acid generation was achieved using commercial iodonium or sulfonium salts (
Scheme 4b).
2. Materials and Methods
2.1. Materials
Diglycidyl ether of bis-phenol A, DGEBA (DER 330, Dow Chemical) was the base model epoxy resin used in this study. The onium salt initiators for EB initiation were cumyl-tolyl-iodonium tetrakis(pentafluorophenyl)borate (Bluesil PI2074, Bluestar Silicone), named DAIS-B and triarylsulfonium hexafluorophosphate (TAS-SbF6) and hexafluorophosphate (TAS-PF6) from Sigma-Aldrich. For UV curing at 385 nm, TAS-SbF6 was used for direct photoactivation. The desired amount of initiator was added to the resin and cautiously dissolved by mechanical stirring under exclusion of visible light. The samples were protected for moisture sorption at the different stages of their preparation.
Benzyl alcohol (BzOH), phenyl glycidyl ether (PGE) and poly(tetrahydrofuran)-diol (number-average MW = 250) and Luperox P were obtained from Sigma-Aldrich, France. PGE was stored over molecular sieves to minimize its moisture content. Unmodified silica powder (grade Millisil E6 with characteristic particle size < 150 μm) was obtained from Sifraco (Entraigues, France) and used as an inert filler after extensive dehydration for 24 h at 90 °C in a ventilated oven for few test samples, as detailed in the section Results and Discussion. In these composite samples, the silica content was 60 wt-%. Poly(ethersulfone) with the reference PES 3600P (low molecular weight, reduced viscosity 0.36 at 1% w/v in dimethylformamide) or PES 5003P (higher molecular weight, reduced viscosity 0.50 at 1 % w/v in dimethylformamide) from Sumitomo Chemical (Japan) was used as a toughening agent dissolved in the precursors of the epoxy matrices.
2.2. Curing by Irradiation
Unless otherwise stated, blends of epoxy resin, onium salts and selected additives were mixed in proportions expressed as wt-% of the final mixture.
Simple bar samples of controlled dimensions (2 mm thick and 5 mm broad, 40 mm long) were obtained in silicon molds. Larger plates were prepared in aluminum molds assembled from 5 mm thick metal plates (from 10 cm × 10 cm to 20 cm × 30 cm) coated with a thin layer of demolding agent and placed on both sides of a 1 to 4 mm thick rectangular metal spacer. The resin was poured into the molds through a special opening on the top surface and then degassed under vacuum (100 mbars, 80 °C). The samples were exposed with their largest dimension perpendicular to the beam of a pulsed electron accelerator (10 MeV Circe II Linac, 20–30 kW) operated with a time-averaged dose rate of ca. 15 kGy s−1 (Ionisos facility, Chaumesnil, France). The dose was adjusted by changing the number of passes applied to the sample at 10, 25 or 50 kGy per pass, as indicated in the text.
In some experiments, a Cu-constantan thermocouple was placed at the center of the sample to record the temperature profile in the resin during radiation processing.
Post-thermal treatments were performed in ovens set at the desired temperature.
2.3. Infrared (NIR) Monitoring
The extent of polymerization was determined by near infrared (NIR) spectroscopy performed with a Vertex 70 spectrometer (Bruker), based on transmission spectra recorded between 8000 and 4000 cm
−1 with a spectral resolution of 4 cm
−1. The decay of the epoxy band vibration at 4529 cm
−1 was measured and normalized to the 4624 cm
−1 aromatic C-H combination band. Monomer conversion (π) was calculated according to Equation (1) [
22]:
Each measurement was triplicated by changing the position of the sample’s probed zone. Conversion values were calculated from the resulting average.
2.4. Structural Characterization of Cured Samples
The monomer conversion (π) was determined by near infrared spectroscopy performed with a Vertex 70 spectrometer (Bruker). Temperature-modulated differential scanning calorimetry (TMDSC) was performed on 5–10 mg samples of cured materials with a TA Instruments Q100 upgraded with the temperature-modulation option. Measurements were conducted from 30 °C to 250 °C, at 1.5 °C·min−1 with a modulation amplitude of 0.5 °C·min−1 and a 90 s period. The glass transition temperature was determined from the position of the maximum of the reversing heat capacity derivative.
The thermomechanical properties were determined with a TA Instruments Q800 Dynamic Mechanical Analyzer. The analyses were carried out on cured sample rods (17.5 mm long, 10 mm broad, 2 mm thick) with a single cantilever clamp at 1 Hz, a 30 µm amplitude and a 3 °C·min−1 ramp temperature.
Micrographs revealing the surface morphology of cryo-fractured samples were recorded using a JEOL7900F scanning electron microscope operated at 1.5 kV.
2.5. Tensile Properties of Cured Samples
K
IC measurements were performed using the test methods of ISO 13586 with single edge notch bend specimen dimensions, 52.8 mm × 6 mm × 12 mm, that was notched and pre-cracked according to the standard. Specimens were submitted to three-point bending test using a Zwick 1485 machine (5 tests for each material) [
23].
3. Results and Discussion
We present here an overview of representative results obtained by curing radiation-curable formulations based on the aromatic epoxy resin DGEBA with an n value close to zero, whereas in practice formulations containing DGEBA oligomers, epoxynovolak (EPN) and solid epoxy prepolymer DEGEBA-Novolak-type, can be formulated to adjust the initial viscosity, reactivity and modulus of the cured materials (
Scheme 4a). In situ acid generation was achieved using commercial iodonium or sulfonium salts (
Scheme 4b). To adjust the reactivity and the cross-link density in the obtained networks, a monoepoxide, phenylglycidyl ether (PGE), as well as transfer agents such as benzylalcohol and poly(tetrahyrofuran)-diol (poly(THF)), were introduced in variable amounts into some formulations. To evaluate the reactivity and the thermophysical behavior of toughened matrices, a high-Tg thermoplastic, poly(ethersulfone) (PES), of limited molecular weight was dissolved into specific blends, whereas a powdered silica was used as mineral filler to perform preliminary tests on model composite materials.
3.1. Monitoring of Epoxy Polymerization
The extent of polymerization was determined at different stages of the curing process by near infrared spectroscopy. A representative series of spectra is shown in
Figure 2a. The measurements performed on DGEBA samples containing various amounts of DAIS-B and cured with two passes of 50 kGy can be compared to a spectrum of the uncured resin of the same thickness (
Figure 2b). Using Equation (1), the conversion levels of epoxy groups were as high as 85, 88, 89 and 95%, for 0.125, 0.25, 0.50 and 1.00 wt-% of iodonium salt, respectively. These results can be compared to the values measured from samples prepared with sulfonium TAS-PF
6 and TAS-SbF
6, with the same concentration range and same radiation treatment of an overall dose of 100 kGy (
Figure 3). The two triaryl sulfonium salts appear much less efficient than DAIS-B. This is due to the decrease in reduction of potential properties of sulfonium (E° (Ar
3S
+/Ar
2S + Ar
•) ≃ −1.2 V vs. SCE) compared to the iodonium cation (E° (Ar
2I
+/ArI + Ar
•) ≃ −0.8 V vs. SCE) [
24,
25].
The strong influence of the nature of counter-anion, hexafluoroantimonate or hexafluorophosphate is clearly evidenced from the comparison between the two salts with the same sulfonium cation [
26]. This confirms the utmost importance of the reduced nucleophilic character which limits ion pair interactions and favors propagation. For all systems, a common trend is the favorable influence of the absorbed dose and the initiator content, which lead to an expected increase of active center concentration, with a direct and positive effect on conversion. Thermal effects resulting from polymerization exothermicity, about 80 kJ · mol
−1 for epoxy monomers, are also considerable in the rapid polymerization of large quantities of bulk monomers [
27,
28].
3.2. Thermal Effects upon Irradiation
To apprehend more precisely the thermal effects associated with the so-called “room temperature” polymerization induced by radiation, thermocouples were placed in resin samples containing 0.5 wt-% of DAIS-B [
29]. The 3 cm thick bulk sample with its 280–300 g of epoxy resin was conveyed under the electron beam to absorb 25 kGy per pass. With an initial energy of 10 MeV, the electron flux goes through the sample with very limited attenuation, as illustrated by the plots of
Figure 1. Energy deposition can be considered to be essentially uniform.
The recorded temperature profile (
Figure 4) shows that after a short period of latency, during which the sample is warmed up by conversion of the incoming beam into heat while the first acidic species to be generated in situ are quenched by basic contaminants and moisture, the temperature in the irradiated sample rises up to 180 °C within about 10 s, the peak being observed as the sample has already moved away from the irradiation zone.
During the second and third pass, the temperature rise is limited, being only due to the absorption and conversion of radiation. Polymerization proceeds in almost adiabatic conditions. Polymerization seems to have been driven to completion. The kinetics of energy dissipation can be appraised from the very smooth slope of temperature decrease 150 s after the onset of irradiation.
More complex temperature profiles have been recorded during other experiments conducted with various contents in onium salt. The occurrence of vitrification between two consecutive passes may occur and block further reaction, resumption of the exposure to radiation being almost inefficient in a glassy matrix even if large amounts of unreacted epoxy functions are available for polymerization. Conversion measurements at various stages of the curing process are expected to bring additional information.
3.3. Dose Effects Combined and Thermal Treatments
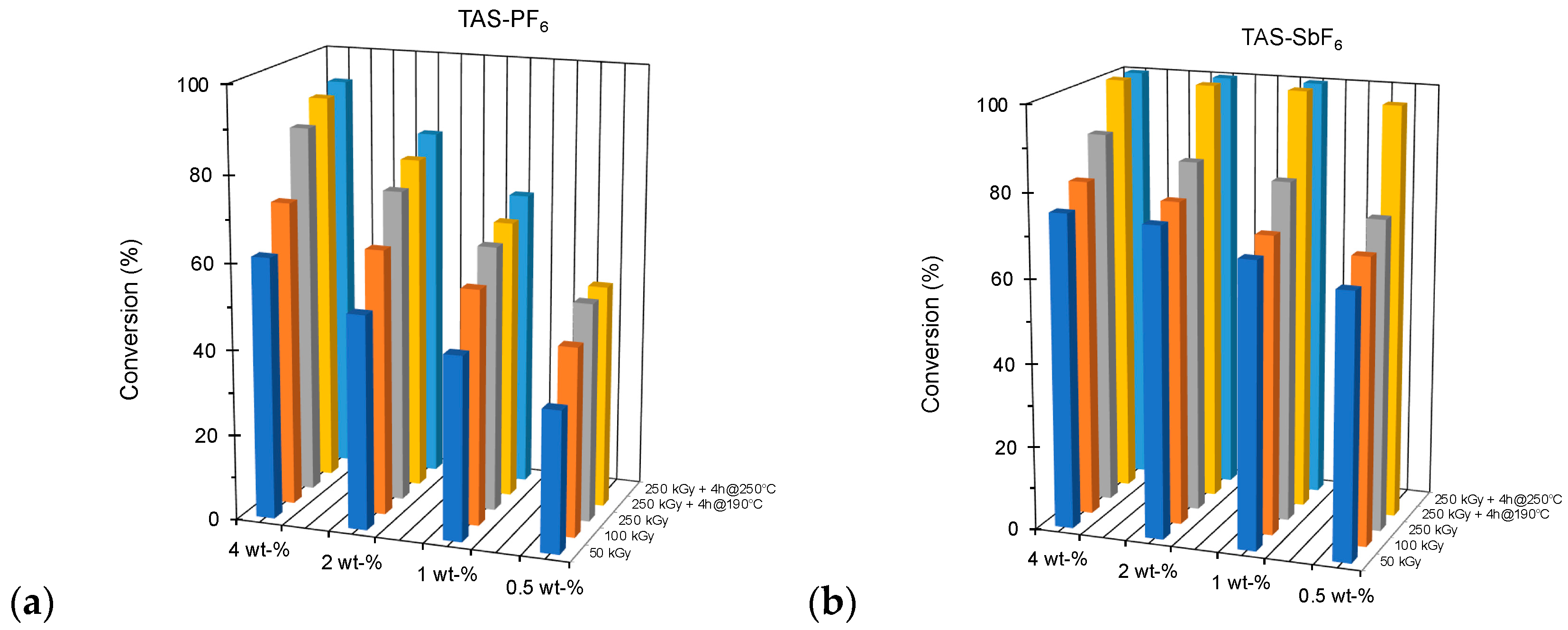
Using two initiators with a smoother reactivity than DAIS-B, we prepared and cured under various conditions two series of blends containing from 0.5 to 4 wt-% of sulfonium salts either with the PF6− or AsF6− counter-anion. Polymerization was induced by EB irradiation at 50 kGy, 100 kGy, and 250 kGy with optional thermal post-treatment at 190 or 250 °C for 4 h.
The results represented in the bar graphs of
Figure 5 confirm without surprise the higher efficiency of the hexafluoro-arsenate salt. It is worth noticing another important difference with respect to the combined effects of initiator concentration and strength of the curing treatment. The comparison is very instructive since
Figure 5a clearly shows that for a given content in onium salt, the harsher the treatment, the lower the increase of conversion, even if the content in unreacted epoxy groups is high. Here, experiments conducted at 190 and 250 °C in the presence of low or even high concentrations of hexafluorophosphate initiator allow to exclude the occurrence of vitrification phenomena between the stages of a curing treatment. This raises questions about the real living feature of the active centers in the presence of PF
6− anions.
Contrastingly, with the hexafluoro-arsenate anion (
Figure 5b), the rougher the treatment and post-curing were, the higher the increase in conversion. It seems that we are here benefiting from the accumulation of active centers and overcoming vitrification without significant loss of chemical reactivity.
3.4. Material Properties
The correlation between epoxy conversion and thermophysical properties of the cured matrices is the articulation between molecular structure and the solidification or hardening of reactive epoxy blends subject to polymerization. The thermomechanical spectra of
Figure 6a recorded during the first ramp of the DMA analysis show the strong difference between the matrix cured at 10 kGy and at the two higher doses (two times at 25 kGy and four times at 25 kGy). The primary observations are explained by the progress of epoxy polymerization at higher doses or by the resumption of pseudo-living cationic polymerization while the thermal ramp of DMA analysis crosses the glass transition of the analyzed sample.
A slight decrease in the temperature of tan δ maximum from 190 °C to 188 °C between the first and the second run is observed for samples cured at 50 and 100 kGy (
Figure 6b). Such a behavior, systematically observed in cured samples, has been discussed already in terms of relaxation of ion pair clusters [
28]. There is here an additional observation with the very interesting data on the evolution of the width of the loss factor recorded during the first run between the 10 kGy irradiated sample and the one cured at higher doses, an indication of the highly heterogeneous distribution of the cross-link density in the network. Moreover, in the sample cured at the lower dose, the right-hand side of the loss factor peak is shifted towards lower temperature, a feature which does not appear in the networks formed from aromatic diacrylates. This is another supporting argument on the reversible formation of domains with higher rigidity under non-equilibrium conditions, likely through additional interchain bridges involving ionic interactions. At the second run, the relaxation profiles are very similar, and subject to possible small artefacts associated with the clamping of specimens in the measuring cell which might explain the small differences in absolute E′ and E″ moduli.
We compared different methods for determining the glass transition Tg of the epoxy networks, either by differential scanning calorimetry (DSC) or by observation of the α relaxation revealed in the spectra recorded by dynamic mechanical analysis (DMA). In DMA analysis, the thermomechanical relaxation is observed with the onset of the steep decay of the storage modulus E′, the maximum of the loss modulus E″ or on peak of the tan δ.
The plots of
Figure 7a were obtained from samples prepared from DGEBA and increasing amounts of TAS-SbF
6. The values determined on the basis of the tan δ values are typically 20 °C higher than those obtained by calorimetry or by the maximum of E″ in DMA spectra, these values being very close for a given material, except for samples of lower conversion and broad distribution of the loss factor, i.e., for highly heterogeneous distribution of cross-link density.
This comparison was also applied to composite samples prepared with the same amount of TAS-SbF
6 (0.25 wt-%) and silica powder as an inert filler (60 wt-%) but various contents in benzyl alcohol as a transfer agent. The decrease in Tg shown in
Figure 7b confirms the efficiency of benzyl alcohol to decrease the cross-link density within the cured networks.
The amount of iodonium salt DAIS-B has a limited effect on the Tg determined as the temperature of the E″ maximum (
Figure 8a). One can, however, observe a slight decrease in Tg for the four higher concentrations in initiator and harsher curing conditions after 100 kGy irradiation. This can be explained at least in part by a relaxation of ionic interactions produced during the very fast occlusion of active centers and vitrification during radiation-initiated curing. The situation is slightly different with the less reactive TAS-SbF
6 initiator (
Figure 8b). For the two lowest contents in sulfonium salts, the extent of curing after irradiation at 100 kGy has obviously not reached its maximal value. Thermal post-curing during the DMA analysis by thermal treatment in an oven allows to approach completion. Interestingly, the observed Tg values for 1 wt-% of TAS-SbF
6 leads to the highest Tg value observed within these two series.
The need for a precise control of Tg in specific applications prompted us to further examine the effect of transfer agents in pure matrices.
3.5. Internal Plasticization Using Reactive Additives
The presence of hydroxyl groups as impurities or as additives impacts the curing kinetics and, among other features of the material, the network Tg. Chain transfer, which occurs by the AM mechanism, leads to shorter polymer chains and increases the mobility of the reactive species. This results in an acceleration of curing, in a higher final conversion of the monomers, and some decrease of Tg. The plots of
Figure 9 illustrate the various possible options to tailor the network structure using the specific reactivity of each type of hydroxyl group [
30,
31,
32]. The use of PGE as a representative reactive diluent to facilitate the mixing or impregnation processes in composite fabrication also has an influence on the Tg of the network [
33].
3.6. Heterogeneities
The curing scenario proposed to describe the gradual build-up of networks by radiation-initiated chain polymerization multi-epoxides is represented in
Figure 10. The progress of DGEBA polymerization illustrates the structural variations at the origin of the increase in Tg and the broadness of the transitions due to the formation of a variety of defects and heterogeneities in cross-link distribution within the networks [
34,
35,
36].
We have already reported on the importance of these structural defects at the origin of the brittleness of networks obtained by fast chain polymerization of multifunctional monomers, in particular in aromatic diacrylates [
37,
38,
39,
40].
Figure 10.
Sketch representing the gradual formation of a monolithic nano-heterogeneous network upon radiation-initiated cationic polymerization DGEBA-based formulations as supported by AFM imaging [
22,
41].
Figure 10.
Sketch representing the gradual formation of a monolithic nano-heterogeneous network upon radiation-initiated cationic polymerization DGEBA-based formulations as supported by AFM imaging [
22,
41].
We recently reported on the advanced understanding of the build-up of thermomechanical properties of networks at nanoscale by probing with new AFM techniques and measuring the peak normal forces, which allow to quantify the local modulus, adhesion, dissipation, and deformation, while simultaneously imaging sample topography of cationically-cured DGEBA and epoxy Novolak resins [
22,
41].
These observations raise the need for progressing in the control of nanoscale morphology of these different types of networks to improve mechanical properties.
3.7. Matrix Toughening by Polymerization-Induced Phase Separation
We prepared simple formulations with 10 wt-% of PES 5003P with respect to the total amount of matrix precursors and a composite with powdered silica representing 60 wt-% of the whole material to examine the effects of this additive on the cationic polymerization of epoxides. Monomer conversion was measured at various moments of the UV-initiated polymerization of these blends containing 1 wt-% of TAS-SbF
6 with respect to the matrix. The polymerization profiles of
Figure 11 show that there is no significant impact on polymerization kinetics, only a possible screening effect due to UV absorption of UV photons by the PES, but no chemical inhibition, no retardation, and only a slightly slower polymerization rate in the presence of silica powder.
We have therefore explored the possibility to introduce a PES thermoplastic toughening agent into the EB-curable epoxy blends. This approach has marked differences with respect to the conventional toughening methodology applicable to heat-curable systems. The thermodynamics and kinetics of the two modes of curing are strongly different, being extremely brief and starting from RT for radiation processing, and slow and at high temperature for thermal curing. These distinct features are expected to favor the early occurrence of thermoplastic demixion upon radiation-induced polymerization and should limit the growth of the nucleated thermoplastic domains if the system starts vitrifying in the early stage of curing. This combination of favorable factors proved to be particularly efficient for designing acrylate-based radiation-cured matrices and composites [
42,
43,
44,
45].
To compare materials prepared by the two curing process, we implemented the method utilizing a tandem initiator system with a peroxide and an iodonium salt, as presented earlier in
Scheme 3. Two series of DGEBA formulations were prepared with poly(THF)-diol as a transfer agent (DGEBA/poly(THF), 93.3:6.7
w/
w), various amounts of PES 3006P (0, 7.5, 10, 12.5, 15, 17.5, 19 wt-%) and either 0.5 wt-% DAIS only as the initiator for EB curing, or 0.5 wt-% DAIS and 0.125 wt-% Luperox P for the thermally-induced polymerization [
46]. Samples cured in the form of 1 mm thick plates by both types of activation (for 90 min at 120 °C plus 60 min at 170 °C, or EB dose of 50 kGy) showed high conversion levels (>95%). Comparison of the thermomechanical spectra recorded by DMA analysis of the two series of samples revealed differences that can be interpreted in terms of distinct types of morphology in materials of similar compositions but subject to different curing modes.
We focus on the plots representing the variations of the loss factor tan δ as a function of the content in PES and of the curing conditions (
Figure 12). In the absence of PES, the epoxy networks present similar DMA spectra, as the peak is centered at 160 °C for both curing methods. For thermally-cured materials (
Figure 12a), the gradual increase of PES in the blend leads to the development of a relaxation peak at 205–210 °C which corresponds to the Tg of pure PES (220 °C), the position of the epoxy network transition being almost unchanged, while the transition associated to PES at higher temperature increases in intensity almost proportionally with the PES content and is shifted towards higher temperatures by 10 °C. The system appears as composed of two coexisting phases and with minor cross-solubility. On the contrary, in EB-cured samples (
Figure 12b), the transition associated with the epoxy network has its maximum shifted to higher temperatures while the distribution of relaxation temperatures is widened, suggesting the incorporation of a significant amount of PES in the network, with possible formation of a mixed interphase. The peak corresponding to the PES phase appears at the same temperature (200 °C), yet with an enhanced intensity as the amount of PES increases.
Scanning electron microscopy provides complementary information consistent with the observations made by DMA. The micrographs selected for
Figure 13 were obtained from the cryo-fractured surfaces of bars prepared from the same two families of blends cured by the two modes of activation.
Figure 13a,b shows the fracture profile of the untoughened DGEBA network prepared by EB curing. At low magnification, shear banding is clearly observed together with fragments of ripped material which also appear on the expanded micrograph [
47]. In EB-cured materials containing rather low amounts of PES (
Figure 13c,d), one observes small PES particles or their imprint in the broken matrix. The particles are evenly distributed with a characteristic size between 100 and 300 nm. In the thermally-cured samples, with 7.5 (
Figure 13e,f) or 19 wt-% of PES (
Figure 13i,j), one observes larger spheroidal phase-separated domains (0.5 to 10 μm in size) with multiphase inclusions observable at larger magnification. With a PES content of 19 wt-%, the spheroidal clusters are more dispersed in size than those in the material containing 7.5 wt-% of PES. EB-cured samples present a different morphology which can be assigned to a co-continuous phase-separated matrix [
48]. Such a structure was obtained by a thermal post-treatment of EB-cured samples containing low amounts of PES, as exemplified by the micrographs of
Figure 13c,d for specimens before thermal post-treatment, to be compared to the pictures of
Figure 13g,h obtained from post-cured specimen. Samples with higher contents in PES were obtained directly with the morphology assigned to a co-continuous structure after EB curing (
Figure 13k,l) [
42].
The overall observations can be rationalized by the following explanations. Cured materials obtained from formulations containing various amounts of PES (from 7.5 to 19 wt-%) exhibit a biphasic morphology contrasting with the one observed with the PES-free epoxy matrix. The timescale of the chain polymerization is much shorter than for thermal curing. The expected polymerization-induced phase separation process takes place and further develops differently depending on the kinetics and thermodynamical conditions. Because of the sudden vitrification of the system upon EB irradiation of samples initially treated at RT, mass transport required to ensure the growth of phase-separated PES clusters is rapidly constrained by the surrounding network. Thermal curing over long reaction times allows for the formation of large demixed domains, sometimes exhibiting a complex morphology with multiple inclusions of PES in the matrix forming spheroidal composite domains included in the epoxy-rich continuous matrix. The geometrical features of the phase-separated domains are, however, of the same characteristic dimension as that of the heterogeneities revealed in various types of diacrylate networks. Differences in terms of morphology are strongly known to influence the corresponding toughness level. KIc values vary from 1.3 to 2.2 MPa·m0.5 for samples having a PES content varying from 7.5 to 19 wt-%. Further investigations are in progress to gain more information on the structure, morphology and local composition of the two classes of cured materials. Detailed analysis of fracture profiles would also contribute interesting information on the propagation of cracks depending on the morphology. It must be noted that for thermally-cured samples, the large spheroidal PES inclusions seem not to have strong interactions with the epoxy matrix. The co-continuous structure suggested from the micrographs obtained from EB-cured samples containing PES seems to be more beneficial to the expected toughening effect.
EB-cured matrices prepared with similar compositions in toughened materials show optimized critical stress intensity factors of (K
Ic) 1.8, 1.7 and 1.6 MPa·m
0.5 for DGEBA-PGE (83:17
w/
w) matrices with PES contents of 10, 15 and 20 wt-%, respectively, whereas the K
Ic value is about 0.6 MPa·m
0.5 for the unmodified reference resin cured in similar conditions (
Figure 14). This confirms the possibility to overcome the inherent brittleness of matrices produced by radiation-induced homopolymerization of multifunctional monomers.
4. Conclusions
Over the recent years, the knowledge on EB and X-ray processing of radiation-curable materials has reached key milestones. Kinetic investigations based on spectroscopic and thermophysical methods provide unique sets of data for describing the gradual evolution of materials upon curing and for enhancing the degree of control over of the curing process and over the properties of obtained materials. The present report overviews the recent advances in the understanding of key factors associated with formulation and processing which control the cationic curing of epoxies. A rational approach encompassing basic considerations of radiation physics, radiation chemistry and polymer science led to an experimental investigation where formulation of reactive blends, radiation treatment under well-controlled conditions, and precise monitoring of transformations at molecular, nanoscopic and macroscopic scales provided key information and insight into the relations between chemical composition, optimized radiation treatment and functional performance for the produced materials.
The results presented in this article exemplify a number of formulation and processing options, either based on the chemical composition of the epoxy formulations, particularly the amount and content of onium salts, or on dose effects and thermal post-treatments. Since sufficient thermomechanical properties after extensive curing are required for structural materials, the thermal profile of the reacting medium has to be well-controlled along the whole process to achieve the final degree of conversion and reach the desired glass transition temperature, Tg. In this respect, the intrinsic reactivity of the acidic species generated in situ from the onium salts is a factor of utmost importance. The nature of both onium cation and counter-anion components of these salts has a strong contribution to the observed reactivity.
Based on previous observations of nano-heterogeneities within the formed networks present, we assessed the relevance of one strategy used for high-performance thermosets which consists of adding a high-Tg thermoplastic such as PES to trigger reaction-induced phase separation during polymerization. We evidenced strong differences in terms of matrix morphology depending on the curing method, by thermal or radiation-assisted initiation. Cured materials obtained from formulations containing various amounts of PES (from 7.5 to 19 wt-%) revealed the biphasic morphology differing from that of the PES-free epoxy matrix. Thermal curing over long reaction times allows for the formation of large demixed domains exhibiting a complex morphology with multiple inclusion. Conversely, early formation of nano-sized phase-separated domains during the cross-linking polymerization of epoxy blends results in the formation of matrices exhibiting enhanced toughness, with KIc values higher or equal to 1.6 MPa·m0.5, whereas the KIc value is about 0.6 MPa·m0.5 for the unmodified reference resin. The weaknesses of the nano-heterogeneous epoxy matrix seem to be alleviated by the purposely provoked formation of nano-sized clusters of toughening thermoplastic. Further investigation of these systems is currently underway.
Improvement of matrix toughness was presented here, emphasizing this unique feature of radiation processing to initiate fast reactions in systems placed at RT. Solutions for matrix toughening with higher processability and better acceptability in terms of environmental and sanitary requirements are under development. Interactions between mineral fillers, glass or carbon fibers, reinforcing textiles and the cationic matrix are being worked on with promising findings for manufacturing composites with outstanding properties. The next milestones will consist of aggregating the key knowledge and consolidated technological options for reducing processing costs while enhancing composite performances using this cleaner and more productive out-of-autoclave manufacturing.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}