
Evaluation of OH Radical Reaction Positions in 3-Ring PAHs Using Transition State Energy and Atomic Charge Calculations


Abstract
:1. Introduction
2. Calculation Methods
3. Results and Discussion
3.1. Literature Research on the Radical Reaction Product
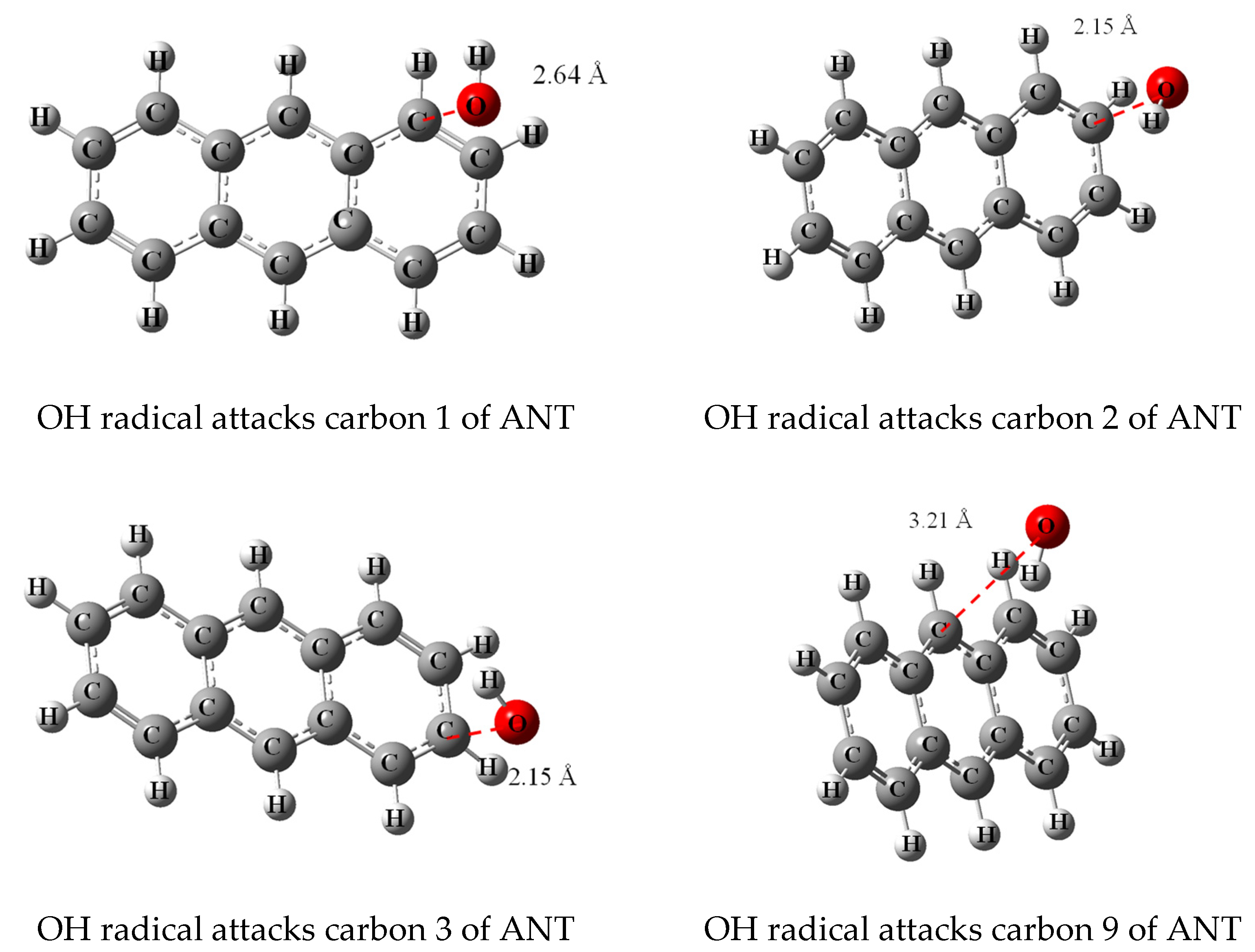
3.2. Transition State Calculation
3.3. Atomic Charge Calculation
4. Conclusions
- The comparison with the test results confirmed that the OH radical reacts at the carbon position with a large negative charge distribution in the carbon ring of the target compound.
- In the case of the target compound, the MK and HLY charges were very accurate in predicting the OH radical reaction position, but the CHelpG charge showed a slight difference.
- In the case of ANT, the carbons at positions 9 and 10 have significantly smaller charge values because they are affected by the benzene rings on both sides. Therefore, it is predicted that the radical reaction at positions 9 and 10 is relatively more favorable than the reaction at other positions.
- The difference between the transition state energy values calculated at all reaction positions of ACEL and ANT was not large, and inconsistent results were obtained even when compared with those of the test results. Therefore, it can be confirmed that the radical reaction position can be sufficiently predicted with the atomic charge calculation only, at a low calculation cost and using a simple calculation method.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Song, M.; Lee, K.; Oh, S.-H.; Bae, M.-S. Impact of Polycyclic Aromatic Hydrocarbons (PAHs) from an Asphalt Mix Plant in a Suburban Residential Area. Appl. Sci. 2020, 10, 4632. [Google Scholar] [CrossRef]
- Achten, C.; Andersson, J.T. Overview of polycyclic aromatic compounds (PAC). Polycycl. Aromat. Compd. 2015, 35, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goswami, A.; Jiang, J.-Q. Comparative performance of catalytic Fenton oxidation with zero-valent iron (Fe(0)) in comparison with ferrous sulphate for the removal of micropollutants. Appl. Sci. 2019, 9, 2181. [Google Scholar] [CrossRef] [Green Version]
- Chu, S.V.; Sands, S.; Tomasik, M.R.; Lee, P.S.; McNeill, V.F. Ozone oxidation of surface-adsorbed polycyclic aromatic hydrocarbons: Role of PAH−surface interaction. J. Am. Chem. Soc. 2010, 13, 15968. [Google Scholar] [CrossRef] [PubMed]
- Kargar1, N.; Amani-Ghadim, A.R.; Matin, A.A.; Matin, G.; Buyukisik, H.B. Abatement efficiency and fate of EPA-Listed PAHs in aqueous medium under simulated solar and UV-C irradiations, and combined process with TiO2 and H2O2. Ege J. Fish. Aquat. Sci. 2020, 7, 15. [Google Scholar]
- Buxton, G.V. Critical review of rate constants for reactions of hydrated electrons, hydrogen atom and hydroxyl radicals (·OH/·O) in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 513. [Google Scholar] [CrossRef] [Green Version]
- Ning, X.; Shen, L.; Sun, J.; Lin, C.; Zhang, Y.; Yang, Z.; Chen, S. Degradation of polycyclic aromatic hydrocarbons (PAHs) in textile dyeing sludge by O3/H2O2 treatment. RSC Adv. 2015, 5, 38021. [Google Scholar] [CrossRef]
- Pérez-Casany, M.P.; Nebot-Gil, I.; Sánchez-Marín, J. DFT theoretical study on the reaction mechanism of the nitrate radical with alkenes: 2-butene, isobutene, 2-methyl-2-butene, and 2,3-dimethyl-2-butene. J. Phys. Chem. A 2000, 104, 10721. [Google Scholar] [CrossRef]
- Yamada, S.; Naito, Y.; Takada, M.; Nakai, S.; Hosomi, M. Photodegradation of Hexachlorobenzene and theoretical prediction of its degradation pathways using quantum chemical calculation. Chemosphere 2008, 70, 731. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.F.; Xu, K.; Lin, M.C. Ab initio kinetics for decomposition/isomerization reactions of C2H5O radicals. ChemPhysChem 2009, 10, 972. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.D.; Iso, M.; Hosomi, M. Prediction of Fenton oxidation positions in polycyclic aromatic hydrocarbons by Frontier electron density. Chemoshpere 2001, 42, 431. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version 6; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A.; Stratmann, R.E.; Burant, J.C.; et al. Gaussian 09, Revision D. 01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Becke, A.D. A new mixing of Hartree-Fock and density-functional theories. J. Chem. Phys. 1993, 98, 1372. [Google Scholar] [CrossRef]
- Breneman, C.M.; Wiberg, K.B. Determining atom-centered monopoles from molecular electrostatic potentials—the need for high sampling density in formamide conformational-analysis. J. Comp. Chem. 1990, 11, 361. [Google Scholar] [CrossRef]
- Besler, B.H.; Merz Jr. K., M.; Kollman, P.A. Atomic charges derived from semiempirical methods. J. Comp. Chem. 1990, 11, 431. [Google Scholar] [CrossRef]
- Hu, H.; Lu, Z.; Yang, W. Fitting Molecular Electrostatic potentials from quantum Mechanical calculations. J. Chem. Theory Comput. 2007, 3, 1004–1013. [Google Scholar] [CrossRef] [PubMed]
- National Library of Medicine. Available online: https://pubchem.ncbi.nlm.nih.gov/compound (accessed on 21 July 2021).


{kind=link}
{kind=link}
| Structure | PAH | Molecular Weight | Boiling Point (°C) | Log Kow * | Vapor Pressure at 25 °C (mmHg) |
|---|---|---|---|---|---|
 | ACEL | 152.2 | 280 | 4.05 | NA ** |
 | ANT | 178.2 | 400 | 4.45 | 1.95 × 10−4 |
| PAH | Reaction Product |
|---|---|
| ACEL | 1,8-naphthalic anhydride |
| ANT | 9,10-anthracenedione |
| Reaction Position | ACEL | ANT |
|---|---|---|
| Ea ** | ||
| 1 | - *** | 117.922 |
| 2 | 45.436 | 116.426 |
| 3 | 45.037 | 116.435 |
| 4 | 45.297 | - |
| 5 | 51.571 | - |
| 6 | - | - |
| 7 | - | - |
| 8 | - | |
| 9 | 118.189 | |
| 10 | - | |
| Position | ACEL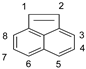 | ANT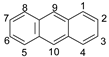 | ||||
|---|---|---|---|---|---|---|
| Charge | ||||||
| MK | CHelpG | HLY | MK | CHelpG | HLY | |
| 1 | −0.295 | −0.215 | −0.259 | −0.254 | −0.223 | −0.247 |
| 2 | −0.295 | −0.215 | −0.259 | −0.127 | −0.060 | −0.107 |
| 3 | −0.256 | −0.249 | −0.251 | −0.127 | −0.060 | −0.107 |
| 4 | −0.092 | 0.029 | −0.050 | −0.253 | −0.223 | −0.247 |
| 5 | −0.305 | −0.317 | −0.315 | −0.253 | −0.223 | −0.247 |
| 6 | −0.305 | −0.317 | −0.315 | −0.127 | −0.060 | −0.107 |
| 7 | −0.092 | 0.029 | −0.050 | −0.127 | −0.060 | −0.107 |
| 8 | −0.256 | −0.249 | −0.251 | −0.254 | −0.223 | −0.247 |
| 9 | −0.353 | −0.440 | −0.466 | |||
| 10 | −0.353 | −0.440 | −0.466 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, M.-J.; Lee, B.-D. Evaluation of OH Radical Reaction Positions in 3-Ring PAHs Using Transition State Energy and Atomic Charge Calculations. Appl. Sci. 2022, 12, 2479. https://doi.org/10.3390/app12052479
Lee M-J, Lee B-D. Evaluation of OH Radical Reaction Positions in 3-Ring PAHs Using Transition State Energy and Atomic Charge Calculations. Applied Sciences. 2022; 12(5):2479. https://doi.org/10.3390/app12052479
Chicago/Turabian StyleLee, Min-Joo, and Byung-Dae Lee. 2022. "Evaluation of OH Radical Reaction Positions in 3-Ring PAHs Using Transition State Energy and Atomic Charge Calculations" Applied Sciences 12, no. 5: 2479. https://doi.org/10.3390/app12052479