1. Introduction
During the past few years, approaches to whole-genome sequencing (WGS) have been evolving [
1]. Since the application of low-throughput first-generation Sanger sequencing, which is definitely expensive and time-consuming, several strategies have been developed [
2]. Among these, next-generation sequencing (NGS) systems, such as IonTorrent, Illumina, or Roche 454, represent low-cost, high-throughput (1.2–6000 Gb) alternatives, but are limited by the reduced length of the produced reads (about 50–600 bp) [
3,
4]. Indeed, short reads often result in an inability to resolve repeat genome regions, leading to misassemblies, gaps, and difficulties in detecting structural variations (SVs), making reconstruction of the original molecules challenging to achieve [
3,
5,
6].
Such an issue has been overcome by the introduction of third-generation sequencing systems, developed by PacBio and Oxford Nanopore Technologies (ONT), able to produce long reads (hundreds to thousands of kb) in a real-time process, resulting in high throughput (20 Gb to 6 Tb), time savings, and cost reductions [
3,
5,
6].
However, long-read sequencing usually presents higher error rates compared to NGS systems (about 15%), although the ONT base-calling tools have been constantly updating to improve accuracy [
7,
8]. Some authors have followed a hybrid approach in de novo assembly, by using short reads from NGS to correct errors in the long third-generation sequences [
9]. Another common approach is to align the reads against a reference genome, an option that is nevertheless not applicable when a high-quality reference is not available in databases [
10].
On the other hand, a weighted bioinformatic analysis might be supportive of error correction, leading to good results [
11]. A key point in gaining an accurate final sequence is the assembly of the reads. In this sense, a hierarchical strategy is one of the best options, consisting of multiple rounds of read overlapping and correction prior to performing assembly [
11,
12]. Several assemblers are available for this purpose, both commercial and open source. Commercial software is intended as user-friendly, stand-alone suites that group several bioinformatic tools and are characterized by a highly accessible graphic interface [
13]. Despite the great benefits in terms of ease of use, these suites, often designed as closed systems, do not offer the same flexibility as open-source tools, which allow fine adjustments to processing parameters in order to adapt to specific requirements or user needs.
Among the open-source assemblers, Canu is one of the most accurate hierarchical tools [
14,
15]. Specifically intended for third-generation sequencing systems, Canu performs correction, trimming (removing adapters and breaking chimeras), and assembly of reads in consecutive steps, while also offering the advantages of a lower runtime and coverage requirements, therefore improving assembly continuity [
12]. This software integrates the MinHash Alignment Process (MHAP), conceived specifically to handle repeats among long-sequencing reads [
12,
16]. Usually, assemblers work by comparing individual
k-mers to identify potential overlaps, resulting in a high computational cost, and by ignoring highly repetitive
k-mers, thus increasing the probability to discard some correct overlaps [
16]. Conversely, Canu adopts a probabilistic algorithm by applying the term frequency and the inverse document frequency to weight and compare compressed MinHash sketches, reducing the chance to select repetitive
k-mers for overlapping [
12]. Additionally, Canu is one of the most configurable assemblers, with hundreds of adjustable parameters, allowing to perfectly fit each assembly to specific necessities [
15].
This feature is particularly useful when challenging sequencing is performed, for instance when bacterial genomes are under investigation, due to some critical features of prokaryotic organisms, such as high plasticity, presence of plasmids, circular replicons with no defined start/end point [
7,
15,
17]. Anyway, achieving the optimal configurations for Canu to obtain the best results might be a tricky task.
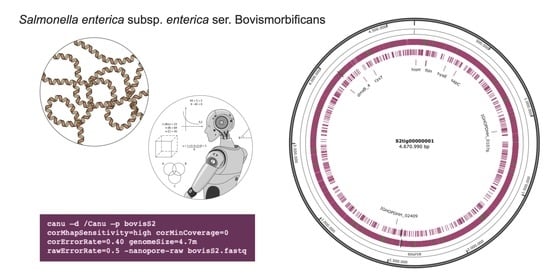
For this purpose, different settings of Canu parameters for de novo assembly and read combinations have been compared, starting from the output of three separate sequencings of a strain of Salmonella enterica subsp. enterica (S.) ser. Bovismorbificans, known for harboring a circular chromosome and at least one large circular plasmid, with the ONT MinION. Therefore, the strain could be considered representative of a large family of bacterial organisms. Additionally, S. enterica serovars are well known and it would be possible to carry out all the appropriate comparisons with the available genome sequences.
The study was carried out on a medium-performance workstation, in order to make such procedures accessible to a wide audience of users. Our results highlighted that the proper setting of Canu may be pivotal in obtaining high-quality assemblies of a bacterial genome that includes a circular chromosome and accessory DNA molecules, such as plasmids.
2. Materials and Methods
2.1. Bacterial Strain and Genomic DNA Extraction
The
S. Bovismorbificans strain used in this study was isolated from the kidney of a pink-backed pelican (
Pelicanus rufescens) living in an Italian zoo, in October 2019. The bird died from chronic kidney failure with gout, and was stored at −80 °C in 15% glycerol within the bacterial collection of the Avian Diseases Unit of the Department of Veterinary Medicine of the University of Bari. The isolation was performed according to the procedures indicated in the terrestrial manual of the World Organization for Animal Health (available online at the address
https://www.oie.int/fileadmin/Home/eng/Health_standards/tahm/3.10.07_SALMONELLOSIS.pdf, latest accessed on 18 February 2022).
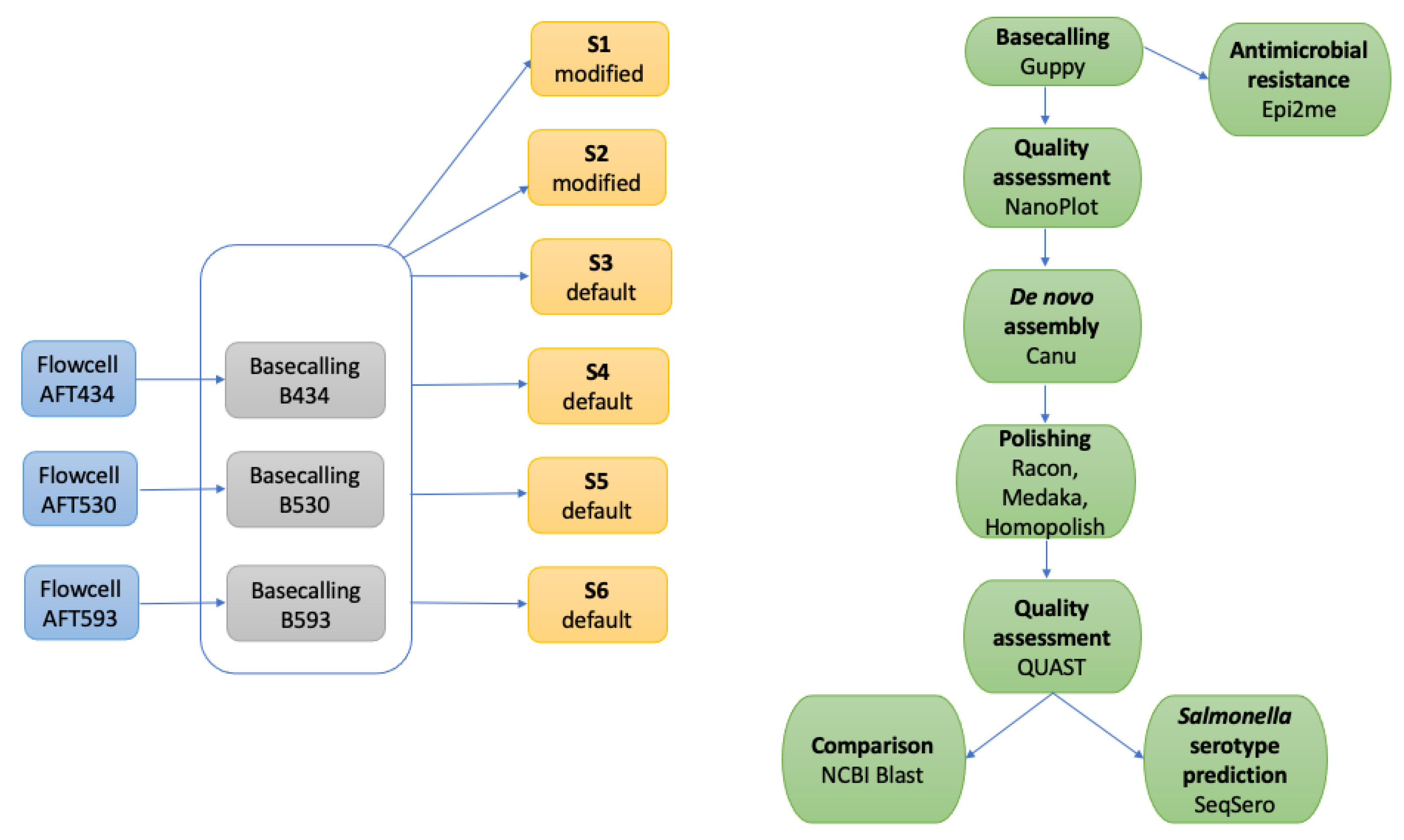
The isolate was revitalized by streaking an aliquot on tryptic soy agar (Thermo Scientific, Milan, Italy), incubated at 37 °C for 24 h. Three separate sequencing experiments were performed from the cultured strain. The workflow scheme has been detailed in
Figure 1.
2.2. Genomic DNA Extraction
Two sequencing experiments were carried out using DNA purified by the Monarch Genomic DNA Purification Kit (New England BioLabs, Frankfurt, Germany), and one with the GenElute Bacterial Genomic DNA Kit (Sigma-Aldrich, Milan, Italy), according to the manufacturer’s instructions.
The recovered DNA was quantified by UV spectrophotometry with Multiskan SkyHigh Microplate Spectrophotometer (Thermo Scientific) and integrity was assessed by electrophoresis on a 1% agarose gel.
2.3. Genome Sequencing
The DNA solutions were used for library preparation with the Rapid Sequencing Kit SQK-RAD004 (Oxford Nanopore Technologies, Oxford, UK) strictly following the manufacturer’s instructions. The sequencing was performed using R9.4.1 flongle flow cells FLO-FLG001 (Oxford Nanopore Technologies) on the ONT MinION Mk1B for 24 h, according to the information provided by the manufacturer (
https://store.nanoporetech.com/eu/flongle-flow-cell-pack.html, latest accessed on 18 February 2022).
The DNA samples were sequenced with three different flow cells, for a total of three consecutive sequencing experiments (named AFT434, AFT530, and AFT593). In particular, AFT434 and AFT530 sequencings were run by loading libraries prepared with 400 ng of the DNA purified with Monarch Genomic DNA Purification Kit, while AFT593 with 400 ng of DNA from the GenElute Bacterial Genomic DNA Kit.
2.4. Basecalling and De Novo Assembly
The basecalling of the raw signals from the three sequencing runs was performed singularly (B434, B530, B593) with Guppy v.5.0.11 (Oxford Nanopore Technologies) by the r9.4.1_450bps_hac model on the ReCaS data center servers (University of Bari). Only the fastq files in the Guppy directory “pass”, considered as high-quality reads, were used for further analyses, singularly and on aggregate (BT).
The quality of the basecalled fastq files from the three sequencing runs has been measured by using NanoPlot v.1.39.0 [
18].
De novo genome assembly of basecalled reads was performed using Canu v.2.2; [
12], with default or adjusted error correction parameters. Combining the DNA extraction methods and the Canu parameters adjustments, six different experimental settings were obtained, as specified in
Table 1.
The correctedErrorRate parameter was decreased from 0.144 (the default value) to 0.12 in S1, while more parameters were modified in S2. Specifically, corErrorRate was modified from 0.30 to 0.04, corMhapSensitivity was set to high, corMinCoverage was adjusted to 0, and corOutCoverage was increased to 100.
The contigs from each assembly setting were polished with one round of Racon v.1.4.10 [
19] and the final consensuses were obtained by Medaka v.1.4.4 (Oxford Nanopore Technologies). Nanopore systematic errors were removed with Homopolish v.0.3.3 [
20].
A comparative de novo assembly was performed using Flye v.2.8.3-b1695 [
21] on the three separate basecalled reads (B434, B530, and B593) and their combination (BT).
2.5. Assembly Quality Assessment
The quality of each assembly was assessed using QUAST v.5.0.2 [
22], comparing them with the
S. Bovismorbificans reference chromosome (GenBank: NZ_CP073715.1, molecule size: 4,667,486 bp). Considering the high variability of the plasmid structures, only the chromosome was considered.
The total number of contigs and bases in the assembly, the length of the largest contig, and N50 (the length for which the collection of all contigs longer than or equal to that value that covers at least half an assembly) were taken into account for each assembly. For the comparison against a reference genome, the number of misassemblies, misassembled contigs, mismatches, and indels were considered.
The nucleotide sequence contigs from each setting underwent a similarity sequence against the nucleotide database of NCBI Basic Local Alignment Search Tool (BLAST) [
23]. Specifically, the smaller contigs were aligned via the web interface, while the larger ones (>1,000,000 bp) with the BLAST+ Command Line Applications tool v.2.12.0 [
24] using the default setting of the task “megablast”.
The
Salmonella serotype determination was performed by using the online versions of SeqSero v.1.0 [
25] and SeqSero2 v.1.1.0 [
26] from the polished assemblies obtained from settings S1, S2, S3, and S6.
Additionally, comparison data from pairwise alignments between each assembly consensus against the reference genome were generated using Artemis Comparison Tool (ACT) v.18.1.0 [
27].
All analyses were performed on a workstation mounting Ubuntu v.20.04.3 LTS (Intel Core i5-4460 3.20 GHz x 4, 8 GB RAM, 1 TB HDD).
2.6. Antimicrobial Resistance Gene Detection
Due to the infectious potential of the sequenced strain, the antimicrobial resistance genes were searched using the ARMA workflow implemented in EPI2ME agent (Oxford Nanopore Technologies).
3. Results
3.1. Genomic DNA Extraction and MinION Sequencing
The DNA extracted in the first two genomic purifications, used in the experiments B434 and B530, was highly concentrated but broken into small fragments. Contrarily, the DNA obtained with GenElute Bacterial Genomic DNA Kit was mostly constituted by high-weight DNA fragments.
Therefore, the MinION sequencing data were extremely variable (
Table 2). In fact, the reads generated from AFT530, where only 33 active pores were detected, were only 21, compared to 116.8 K for AFT434 and 156.76 K for AFT593, which both harbored 75 active pores. Similarly, the N50 from AFT434 and AFT530 was very low, being 873 and 600, respectively, compared to 6.61 K for AFT593. Interestingly, the N50 of AFT434 was low despite the high number of available pores.
The quality values of the basecalled reads are reported in
Table 3.
3.2. De novo Assembly and Quality Assessment
Data about de novo assembly and QUAST quality assessment against the reference genome are reported in
Table 4.
Canu failed in assembling the reads generated from AFT434 and AFT530 (settings S4 and S5) with default parameters due to the extremely high percentage of reads shorter than 1000 bp (90.143% for S4 and 87.9244% for S5).
The best results overall were achieved with setting S2, in which Canu parameters were more finely adjusted. The S2 assembly generated only two contigs (instead of seven for both S1 and S3, and eight for S6), one of them very close in length to the reference chromosome size.
Likewise, S2 exhibited the highest coverage (77.16X), but only minor variations were observed among the other succeeded experiments.
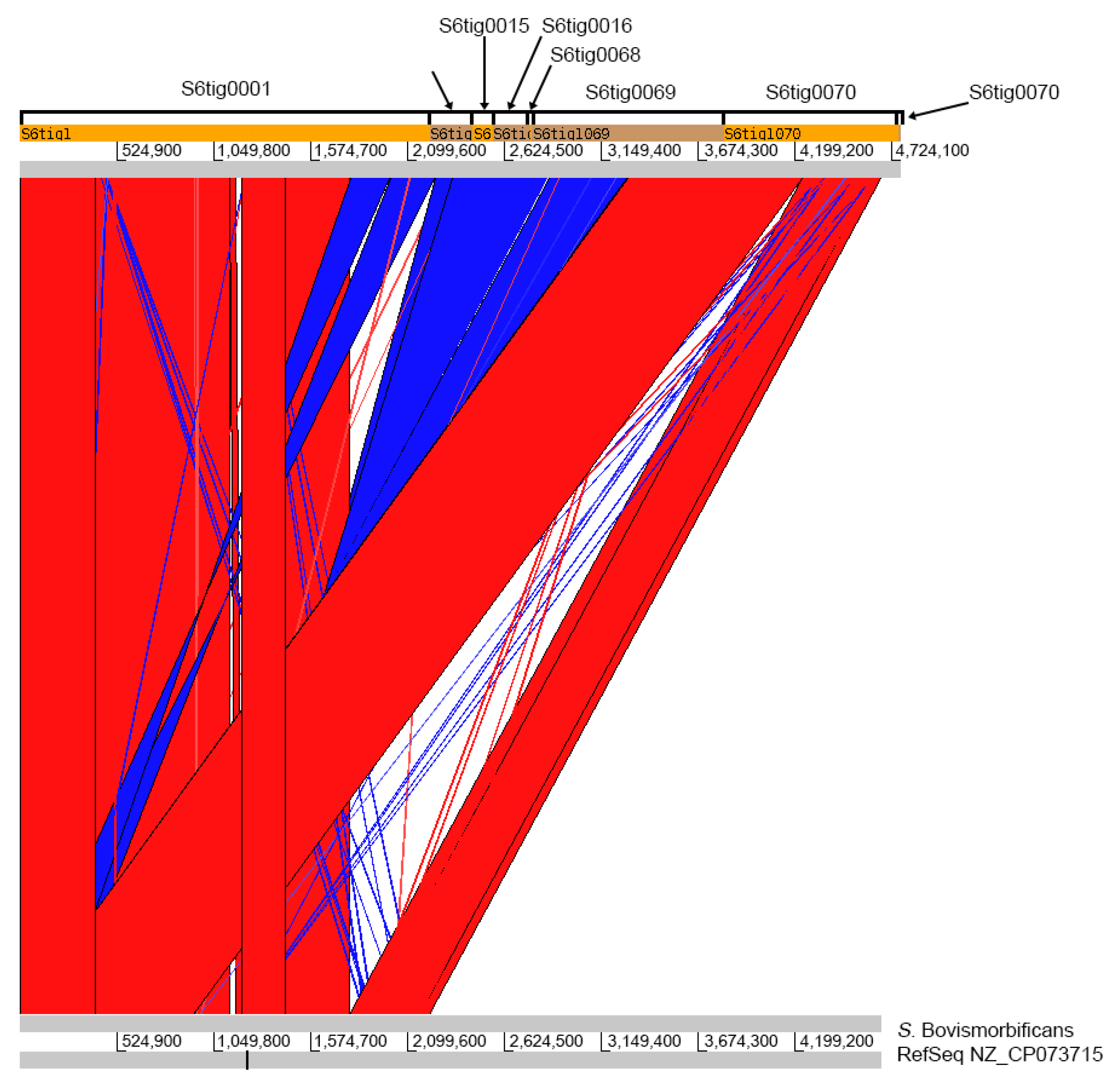
Two misassemblies in only one contig were reported by QUAST in all the settings, with the total number of mismatches ranging from 990 (S1) to 1439 (S6), and indels from 340 (S1) to 759 (S6). Even in this case, the worst results have been observed in the S6 experiment, which consisted of the results of only one sequencing run.
The unaligned contig length was longer in S1 and S3 (183,520 and 184,317, respectively), and shorter in S2 and S6 (153,946 and 154,546, respectively).
The contig S2tig0001, obtained with setting S2, was alignable with the reference genome, and it almost completely covered the S. Bovismorbificans chromosome. For S3, three contigs (S3tig0001, S3tig0011, and S3tig0007) were aligned to the chromosome, with the other four being unaligned. For S6, only one contig (S6tig0015) remained unaligned to the reference chromosome. The shortest number and length of gaps were observed in S2, while the longest was in S6. Overall, only the S2tig0001 extended along the whole chromosome of S. Bovismorbificans, with the exception of some minor gaps.
Gaps retrieved in contigs obtained from all settings are reported in
Table 5 as detected by QUAST.
The total number of gaps varied among the settings, with 6 gaps in S2 (aggregate length 46,474 bp), 7 gaps in S1 (63,995 bp), 7 gaps in S3 (62,538 bp), and 10 gaps in S6 (73,118 bp). Three contigs from S1 were aligned to the reference chromosome (namely, S1tig0012, S1tig0008, and S1tig0001), while the other four were left unaligned. No contig completely spanned the chromosome. Four gap breakpoints were shared among all the obtained contigs. Moreover, S1, S3, and S6 shared another similar gap, and S6 had other two gaps not seen in the other settings. Interestingly, the 30,841 bp and the 13,459 bp gaps correspond to prophage genomic islands in the reference chromosome, therefore regions were characterized by an intrinsic mobility potential. Equally, the 245 bp gap corresponded to another phage gene.
Most of the contigs not aligned to the chromosome were shorter than 400 kbp. Additionally, comparing those data with the sequence similarity searching with BLAST nucleotide database (
Table S1), some of the unaligned contigs matched with
S. Bovismorbificans plasmid.
The serotype prediction obtained by using SeqSero was not sufficient to predict the serovar Bovismorbificans in all the tested assemblies (
Supplementary Results S1). Specifically, SeqSero correctly predicts the serovar only for settings S1 and S2.
Conversely, all de novo assembling with Flye failed, probably because of the poor performance of the available workstation since the tool returned the error “Looks like the system ran out of memory”, indicating the hardware equipment was insufficient for performing the required tasks.
3.3. Detection of Antimicrobial Resistance Genes
No genes related to the antimicrobial resistance were detected, consistently with the antimicrobial susceptibility test that detected no resistance (data not shown).
4. Discussion
The long-read sequencing technology, such as the ONT MinION one, is achieving optimal results in sequencing bacterial genomes [
7]. Although long reads significantly reduce the problem of fragmentation in genome assemblies, contiguity might not be completely ensured. In this sense, the quality of experiments may be severely affected by several factors.
Among these, the quality of the DNA to be included in the library is pivotal. In particular, high molecular weight DNA is fundamental to generate longer, and the here-reported findings strongly support this evidence, considering that the sequencing runs obtained from the two samples characterized by high fragmentation showed a low N50 read length, which made it impossible to proceed with the assembly step.
Another crucial element is the quality of flow cells, in terms of pores available for sequencing. In fact, the worst results were obtained using a flow cell with 33 available pores, probably too few to generate adequate data to be assembled, at least alone. It should be underlined that the manufacturer clearly recommends using only flow cells with more than 50 pores, also offers an efficient warranty policy for cells not complying with optimal features upon arrival to the customer’s facility. Interestingly, the N50 read length obtained from the low-quality cell was only slightly lower than those from the first one, but the overall number of reads was sensibly reduced. This confirms that the number of pores is related to the throughput of the run, while the DNA integrity affects the read length. Therefore, only the combination of high-quality DNA and efficient cells may ensure optimal results.
Nonetheless, particular attention must be always paid to the downstream analysis of the output of the run, especially if de novo assembly is required. In that case, the production of a contig representative of the entire molecule with a coverage high enough to reduce at the minimum the sequencing errors would be desirable. Among strengths, the direct analysis of the potential resistance gene set harbored by a strain has been often appreciated [
28]. Despite the fact that the here-analyzed did not provide the specific resistance gene, the analysis was carried out rapidly and accurately, since the results obtained using the ARMA workflow matched with those from the assembled sequences.
Long-read sequencing systems have been proven to be superior in reaching high quality in the de novo assembly, but some drawbacks might be observed. In fact, Canu, considered the best solution for the de novo assembly of long reads, might fail to produce high-quality contigs if not properly and finely tuned.
Even not considering the assembly of the low-quality outputs (S4 and S5), when Canu was launched with default parameters, the assembly quality was lower in terms of N50 length (3,162,557 for S3, 1,040,624 for S6), number of contigs (seven for S3 and eight for S6) and gap number and length (7 gaps with a total 62,538 bp length for S3, and 10 gaps with a total 73,118 bp length).
Several strategies have been addressed to prevent the failure in reaching full coverage of a bacterial chromosome. The hybrid assembly has been of such approach, using short reads to fill gaps in the scaffold generated by MinION assemblies [
9]. In the present study, the overall output has been enriched by combining the long, high-quality reads of the AFT593 run with the shorter ones from the AFT434 and AFT530 runs. Even with the default setting of Canu, such enrichment has improved, despite marginally, the quality of the contig (S3) if compared to the contig only deriving from the best run (S6).
Better results have been gained when Canu parameters have been adjusted. In setting S1 only one parameter was adjusted, namely the “correctedErrorRate” (the allowed difference in an overlap between two corrected reads; with a default value of 0.144 for Nanopore reads). This value was slightly decreased to 0.12, after considering that coverage for all the settings was more than 72X, as suggested by the Canu developers for high coverage datasets (>60X). In this case, the assembly quality was much better than those obtained in S6, but compatible with setting S3 (N50 length of 3,162,545 bp, seven contigs produced, and seven gaps with a total 63,995 bp length).
More parameters were adjusted in S2. In particular, the “corErrorRate” value (defined as “do not use overlaps with error rate higher than this when computing corrected reads” in the Canu documentation available at
https://canu.readthedocs.io/en/latest/parameter-reference.html, latest accessed on 18 February 2022) was increased from 0.30 to 0.40 to improve the corrected coverage; “corMhapSensitivity” (namely, the “coarse sensitivity level, based on read coverage”) was set to “high”, despite the developers suggested using it for low coverages; “corMinCoverage” (“limit read correction to only overlaps longer than this”) was brought to 0 to correct as many reads as possible; “corOutCoverage” (which controls how much coverage in corrected reads is generated) was set to 100. Conversely, the “correctedErrorRate” was left default in this setting.
That setting (S2) allowed obtaining the best assembly quality. In fact, N50 length resulted in 4,670,990 bp, and only two contigs were generated, long 104,427 bp and 4,670,990 bp, respectively. The BLAST search showed that the shorter contig showed homology with a plasmid of S. Bovismorbificans, while the largest corresponded to the chromosome of the same Salmonella serovar. Artemis ACT showed that such a contig covered the entire S. Bovismorbificans chromosome, without evidencing other rearrangements than the six detected gaps. Three of those gaps were probably due to the actual lack of such regions in the sequenced molecule since they corresponded to prophage genomic islands within the reference sequence. Therefore, the S2 setting was found to be the most appropriate also in terms of number and length of gaps, since only six gaps were detected, with a low probability of being artifactual.
In fact, most of the contig breaks in the bacterial DNA molecules are due to the intrinsic plasticity of microbial genomes. Those genomes are characterized by adaptive modifications (i.e., repeats, insertions or deletions, single nucleotide polymorphisms, inversions) as a result of prophage insertion and/or excision, transposition events, or rearrangements with mobile genetic elements as plasmids, bacteriophages, and transposons [
29]. Usually, long-read sequencing systems can overcome the issue of repeated elements, as their reads may be larger than repeats. Anyway, all those modifications may lead to gaps and misassemblies in de novo assembly. Considering that the largest contig breaks detected in the present investigation are associated with genomic prophage islands, this is another hint about the capability of third-generation sequencing to provide a comprehensive picture of the genomic composition of bacteria. It is noteworthy that the serovar automated prediction returned different results, according not only to the contig quality, but also to the software version, since only SeqSero2 succeeded in determining the serovar Bovismorbificans, and only when the contigs from S2 were computed.
On the other hand, some researchers complain about the longer computational time required for Canu in contrast to other assemblers (personal communications). The Canu documentation warns that the adjustment of some parameters may hugely increase the computational time. However, with a medium-performance personal computer, the assembly took about 13 h with S2, thus confirming that the tools are quite fast in reading assembling. Nevertheless, a reduction in computational time was observed by using MinHash, along with a lower RAM demand [
30].
Instead, they always require several rounds of polishing, which obviously extend the final computational time, despite some Authors obtained good assemblies with only one round of polishing after running of Canu [
12].
In contrast, no assembly was obtained when Flye was used as an assembler. This was probably due to the different algorithms adopted by the two software. While Canu is a hierarchical tool with a probabilistic approach to the alignments of
k-mers, Flye accurately bridges the disjointigs obtained in the initial steps of the computing process [
21]. Despite being less demanding if compared with other assemblers (e.g., Bowtie, BWA, and SOAP2), it still requires high memory usage and, consequently, high RAM availability [
31]. Moreover, some authors found Flye, when used in association with Pilon for the hybrid approach, more inaccurate, with respect to Canu, for producing bacterial genome sequences [
32].
All those considering, the nanopore sequencing with flongle cells may be inexpensive and time-saving and may provide high-quality results, if some conditions are strictly observed. Among those, the good enough quality of the purified DNA, the functional efficiency of flowcells, and a proper setting of the assembly software cannot be superseded.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}