
Line-Shaped Illumination: A Promising Configuration for a Flexible Two-Photon Microscopy Setup



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Featured Application
Abstract
1. Introduction
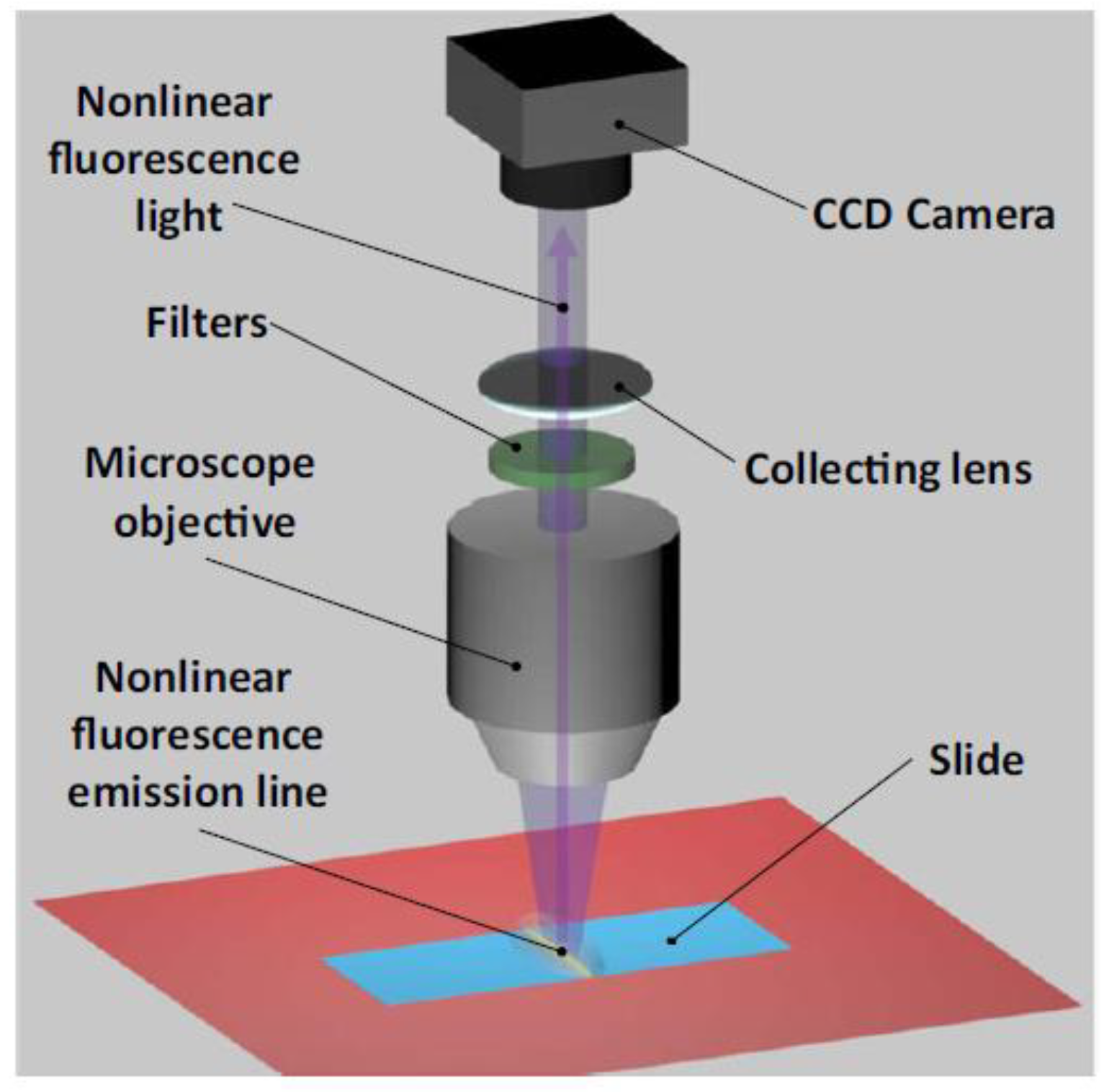
2. Description of the Experimental Setup
3. Overview of Applications
3.1. Two-Photon Spectral Measurements
3.2. Two-Photon Cross-Sections
3.3. Imaging of Biological Samples
3.4. In-Depth Sample Reconstruction
4. Conclusions
5. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- So, P.T.C.; Dong, Y.C.; Masters, B.R.; Berland, K.M. Two-Photon Excitation Fluorescence Microscopy. Annu. Rev. Biomed. Eng. 2000, 2, 399–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denk, W.; Strickler, J.H.; Webb, W.W. Two-photon laser scanning fluorescence microscopy. Science 1990, 248, 73–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berland, K.M.; So, P.T.; Gratton, E. Two-photon fluorescence correlation spectroscopy: Method and application to the intracellular environment. Biophys. J. 1995, 68, 694–701. [Google Scholar] [CrossRef] [Green Version]
- Brakenhoff, G.J.; Müller, M.; Ghauharali, E.I. Analysis of efficiency of two-photon versus single-photon absorption for fluorescence generation in biological objects. J. Microsc. 1995, 183, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Schultz, S.R.; Copeland, C.S.; Foust, A.J.; Quicke, P.; Schuck, R. Advances in two photon scanning and scanless microscopy technologies for functional neural circuit imaging. Proc. IEEE Inst. Electr. Electron. Eng. 2017, 105, 139–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carriles, R.; Shafer, D.N.; Sheetz, K.E.; Field, J.J.; Chisek, R.; Barzda, V.; Sylvester, A.W.; Squier, J.A. Imaging techniques for harmonic and multiphoton absorption fluorescence microscopy. Rev. Sci. Instrum. 2009, 80, 081101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rumi, M.; Perry, J.W. Two-photon absorption: An overview of measurements and principles. Adv. Opt. Photonics 2010, 2, 451–518. [Google Scholar] [CrossRef]
- Guild, J.B.; Webb, W.W. Line scanning microscopy with two-photon fluorescence excitation. W-Pos. 192. Biophys. J. 1995, 68, A290. [Google Scholar]
- Brakenhoff, G.J.; Squier, J.; Norris, T.; Bilton, A.C.; Wade, M.H.; Athey, B. Real-time two-photon confocal microsocopy using a femtosecond, amplified Ti:sapphire system. J. Microsc. 1996, 181, 253–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tal, E.; Oron, D.; Silberberg, Y. Improved depth resolution in video-rate line-scanning multiphoton microscopy using temporal focusing. Opt. Lett. 2005, 30, 1686–1688. [Google Scholar] [CrossRef] [PubMed]
- Parravicini, J.; Tartara, L.; Hasani, E.; Tomaselli, A. Fast calculation of the line-spread-function by transversal directions decoupling. J. Opt. 2016, 18, 075609. [Google Scholar] [CrossRef]
- Tomaselli, A.; Tartara, L.; Hasani, E. Two-Photon Excited Fluorescence Microscope. European Patent WO/2014/154279, 28 March 2013. [Google Scholar]
- Hasani, E.; Parravicini, J.; Tartara, L.; Tomaselli, A.; Tomassini, D. Measurement of two-photon-absorption spectra through nonlinear fluorescence produced by a line-shaped excitation beam. J. Microsc. 2018, 270, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Parravicini, J.; Tomaselli, A.; Hasani, E.; Tomassini, D.; Manfredi, N.; Tartara, L. Practical two-photon-absorption cross sections and spectra of Eosin and Hematoxylin. J. Biophotonics 2020, 13, e202000141. [Google Scholar] [CrossRef] [PubMed]
- Hasani, E.; Tartara, L.; Tomaselli, A. A novel technique for the measurement of twophoton absorption spectra of dyes for nonlinear fluorescence microscopy. In Proceedings of the 2019 IEEE International Conference on BioPhotonics (BioPhotonics), Taipei, Taiwan, 15–18 September 2019; pp. 1–3. [Google Scholar] [CrossRef]
- Hasani, E.; Tartara, L.; Tomaselli, A. Line-scanning two-photon microscope for the investigation of biological samples. In Proceedings of the 2019 IEEE International Conference on BioPhotonics (BioPhotonics), Taipei, Taiwan, 15–18 September 2019; pp. 1–2. [Google Scholar] [CrossRef]
- Pasquetto, M.V.; Santonastaso, A.; Tomaselli, A.; Vaghi, P.; Hasani, E.; Tartara, L.; Scotti, C. Lipoprotein (a) Internalization by RAW 264.7 Cells is Associated with Morphological Changes and Accumulation of Lipids Detectable by Two-photon Scanning Microscopy. Br. J. Med. Med. Res. 2016, 17, 1–12. [Google Scholar] [CrossRef]
- Edelstein, A.D.; Tsuchida, M.A.; Amodaj, N.; Pinkard, H.; Vale, R.D.; Stuurma, N. Advanced methods of microscope control using μManager software. J. Biol. Methods 2014, 1, e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- μManager Official Website. Available online: https://micro-manager.org/ (accessed on 20 January 2022).
- Sutherland, R.L. Handbook of Nonlinear Optics, 2nd ed.; Marcel Dekker: New York, NY, USA, 2003. [Google Scholar]
- Zipfel2020—Two-Photon Action Cross Sections of Alexa 488 Colorant, from Zipfel Lab Online Database of Cornell University. Available online: http://zipfellab.bme.cornell.edu/ (accessed on 10 April 2020).
- Xu, C.; Webb, W.J. Measurement of two-photon excitation cross sections of molecular fluorophores with data from 690 to 1050 nm. Opt. Soc. Am. B 1996, 13, 481. [Google Scholar] [CrossRef]
- Cabrera, R.; Gabriel, M.; Estrada, L.C.; Etchenique, R. Direct Measurement of Two-Photon Action Cross Section. Anal. Chem. 2019, 91, 5968. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parravicini, J.; Hasani, E.; Tartara, L. Line-Shaped Illumination: A Promising Configuration for a Flexible Two-Photon Microscopy Setup. Appl. Sci. 2022, 12, 3938. https://doi.org/10.3390/app12083938
Parravicini J, Hasani E, Tartara L. Line-Shaped Illumination: A Promising Configuration for a Flexible Two-Photon Microscopy Setup. Applied Sciences. 2022; 12(8):3938. https://doi.org/10.3390/app12083938
Chicago/Turabian StyleParravicini, Jacopo, Elton Hasani, and Luca Tartara. 2022. "Line-Shaped Illumination: A Promising Configuration for a Flexible Two-Photon Microscopy Setup" Applied Sciences 12, no. 8: 3938. https://doi.org/10.3390/app12083938