
The Impact of the Temperature Control Strategy in Steady-State Virtual Vacuum Simulation on the Spontaneous Evaporation Rate and Corresponding Evaporation Coefficient


Abstract
:1. Introduction
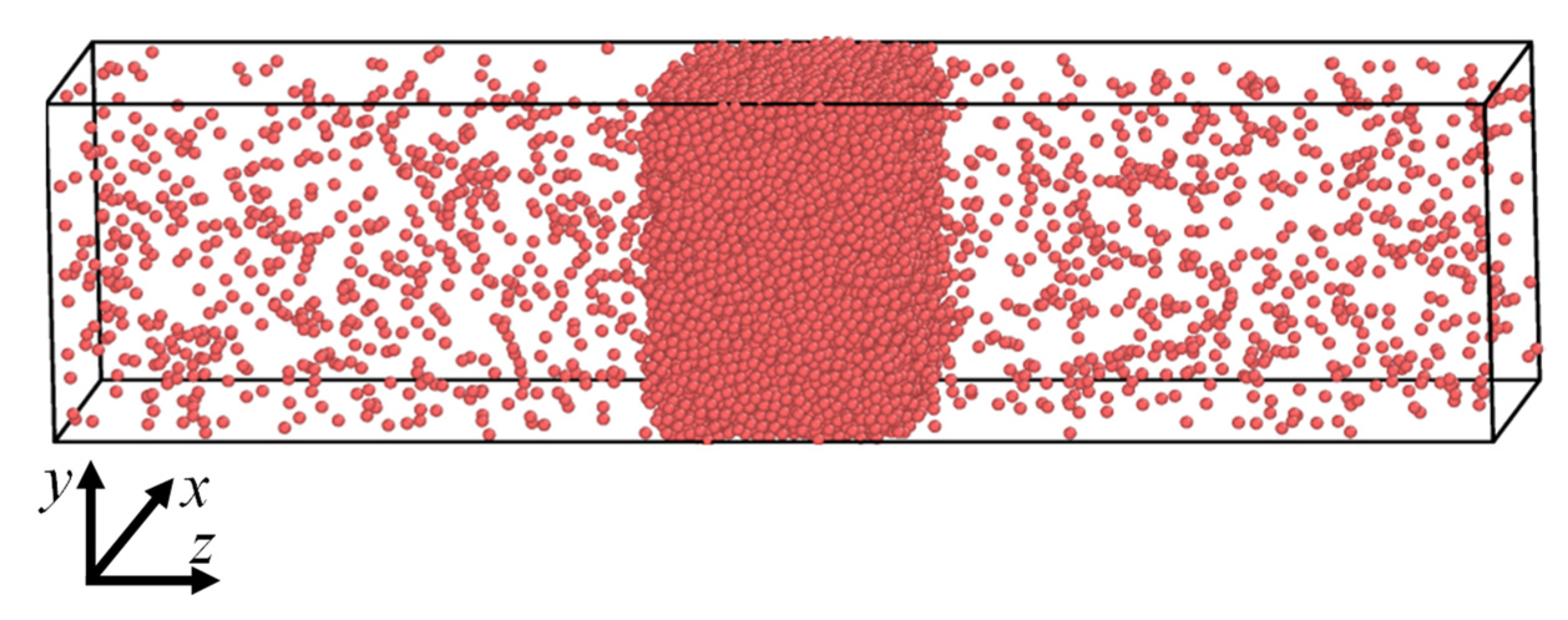
2. Simulation Method
2.1. Liquid-Vapor Phase Equilibria Simulation
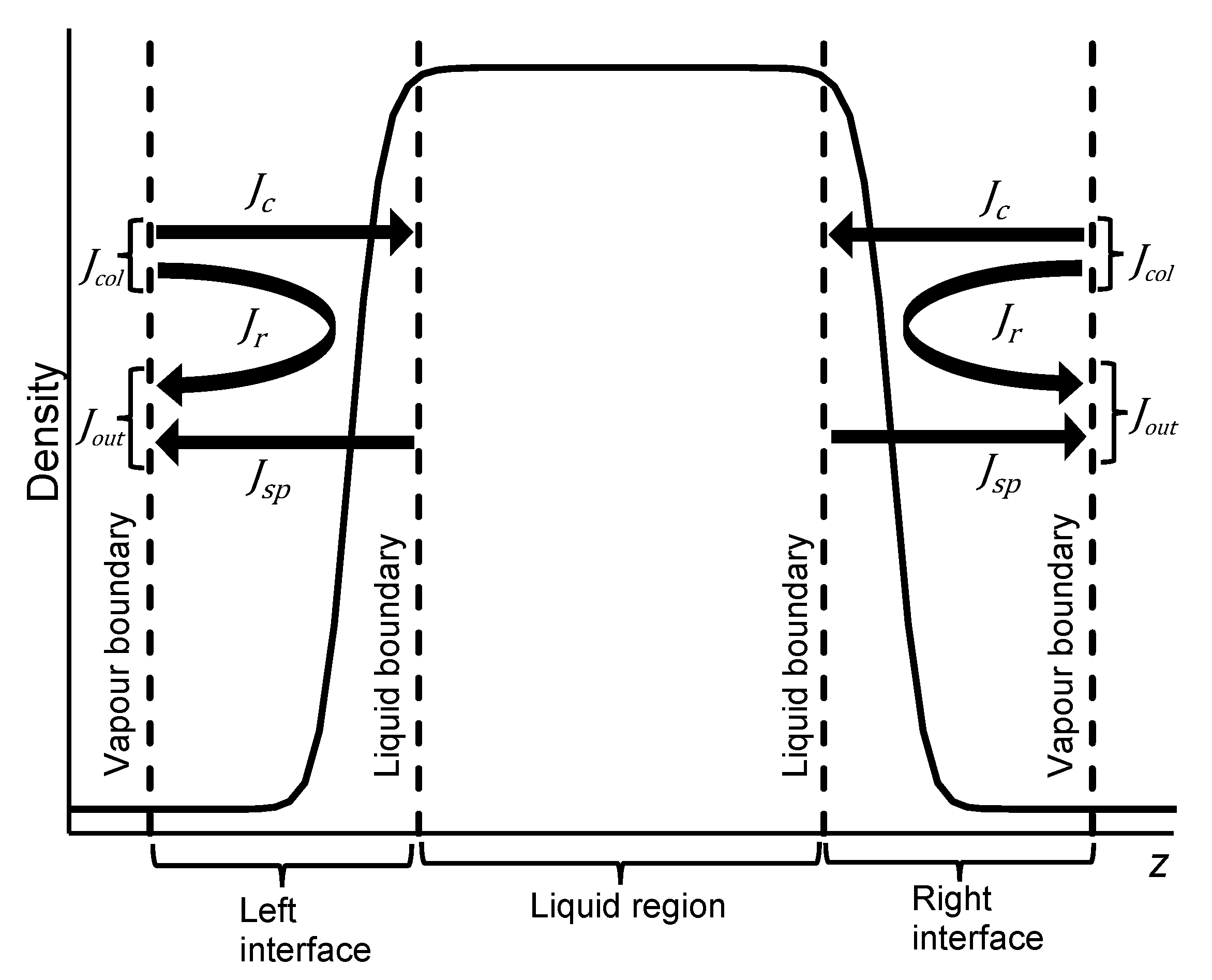
2.2. Deletion Vacuum Simulation
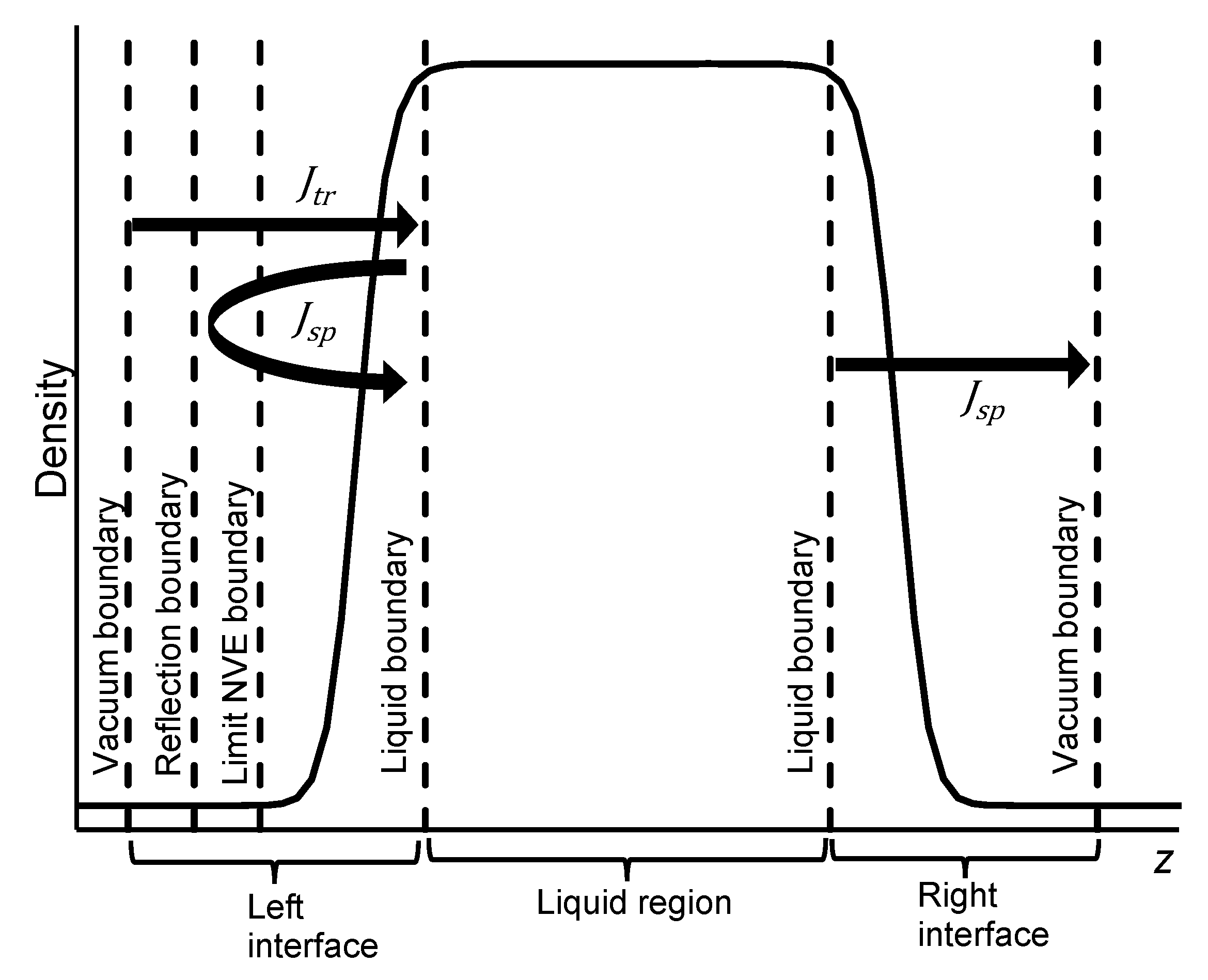
2.3. Steady-State Vacuum Simulation
2.4. Net Momenum in Steady-State Vacuum Simulation
2.5. Simulation Details
3. Results and Discussion
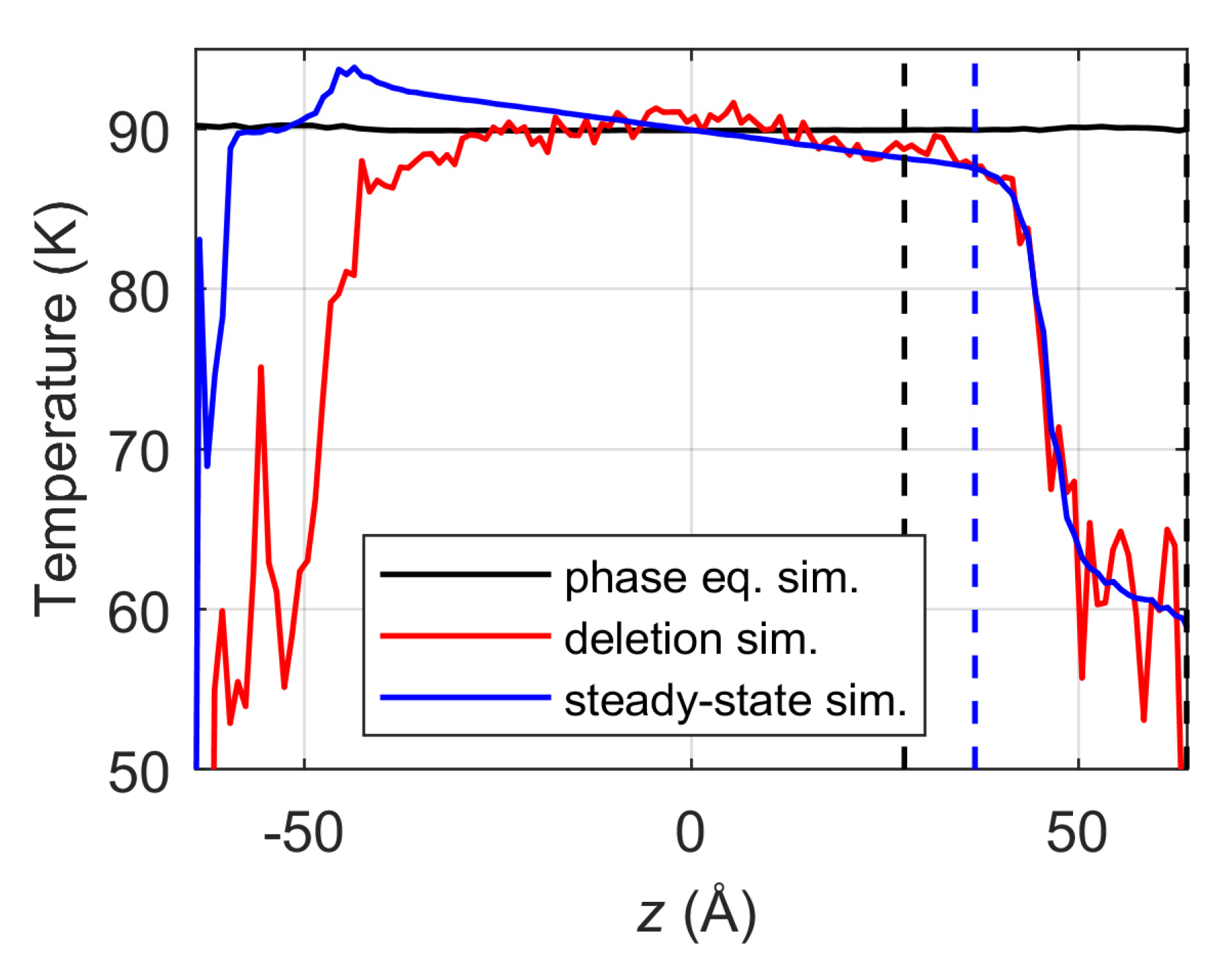
3.1. Temperature Profile in Virtual Vacuum Simulation
3.2. Spontaneous Evaporation Rate Dependency on Surface Temperature
3.3. Velocity Distributions of Evaporating Atoms
4. Conclusions
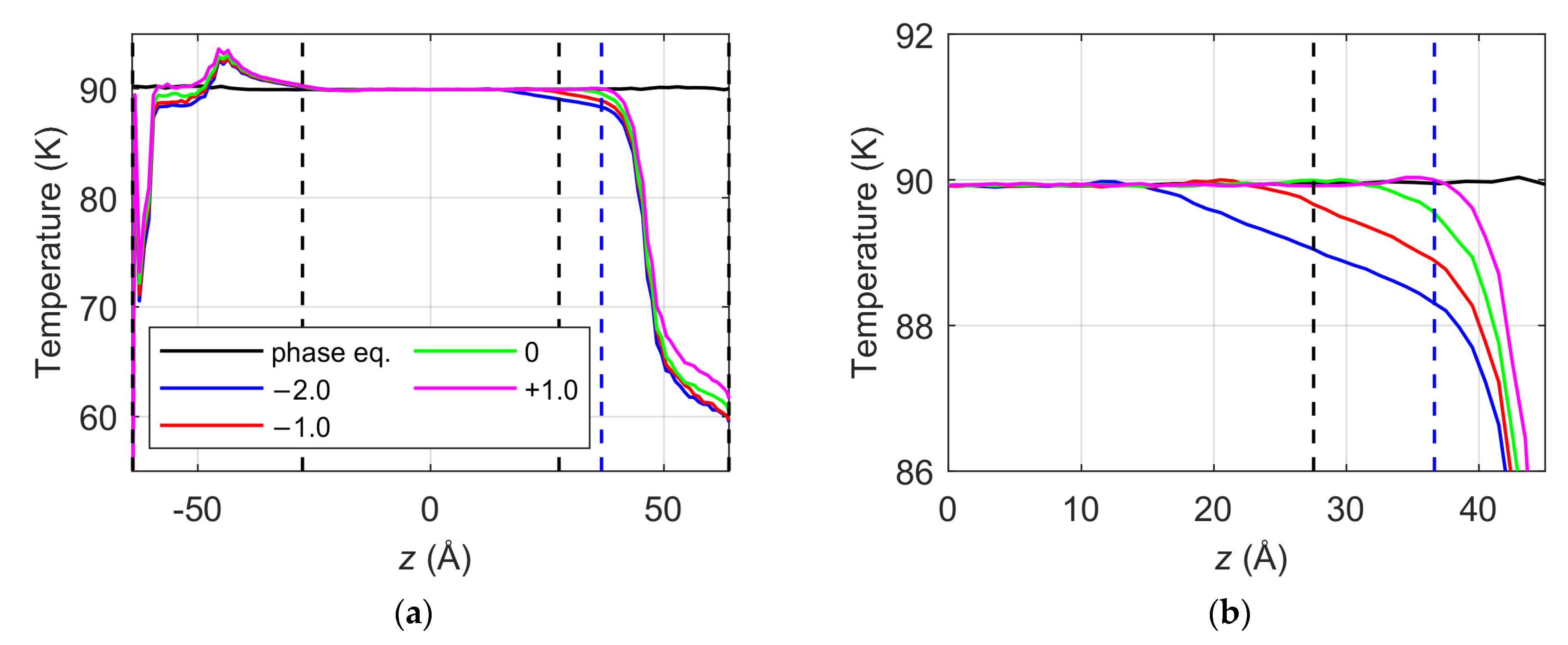
- In both the deletion vacuum and the steady-state vacuum simulations, the temperature control approach, in which the thermostat is simply applied only to the liquid phase and the interfaces are simulated in the NVE ensemble, provides non-uniform parabolic temperature profiles across the liquid phase due to the evaporative heat sink at the evaporating surface. Consequently, the temperature at the liquid surface position is obtained lower by several K compared to the targeted simulation temperature of 90 K.
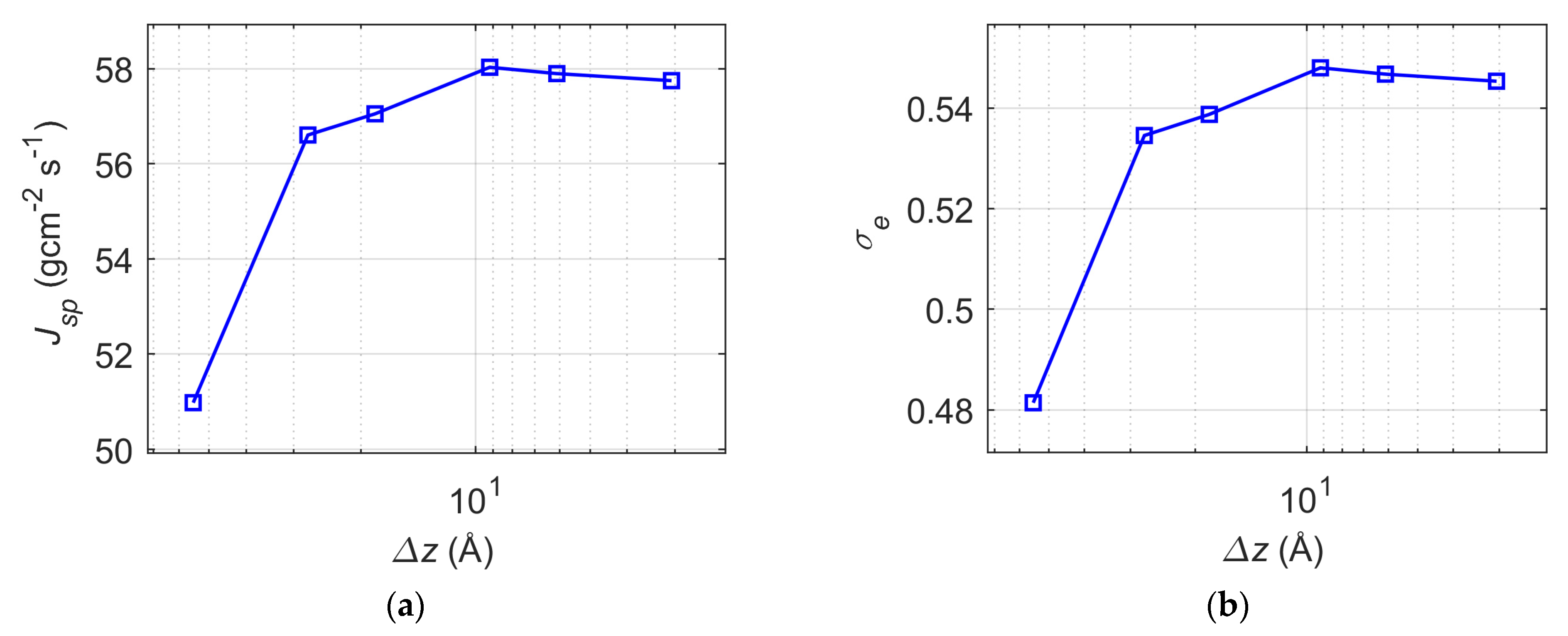
- In steady-state vacuum simulations, the division of the liquid phase into 2 bins, to which the thermostat is applied separately, already flattens the non-uniform temperature profiles and raises the liquid surface temperature; however, the surface temperature reaches saturation value below the targeted temperature with the further reduction of bin width below 9.2 Å. Consequently, the division into 2 bins leads to increased spontaneous evaporation rates, with the evaporation coefficient values increasing by 11%, and the spontaneous evaporation rate increases by another 2.5% and reaches saturation value when bin width is reduced to 9.2 Å.
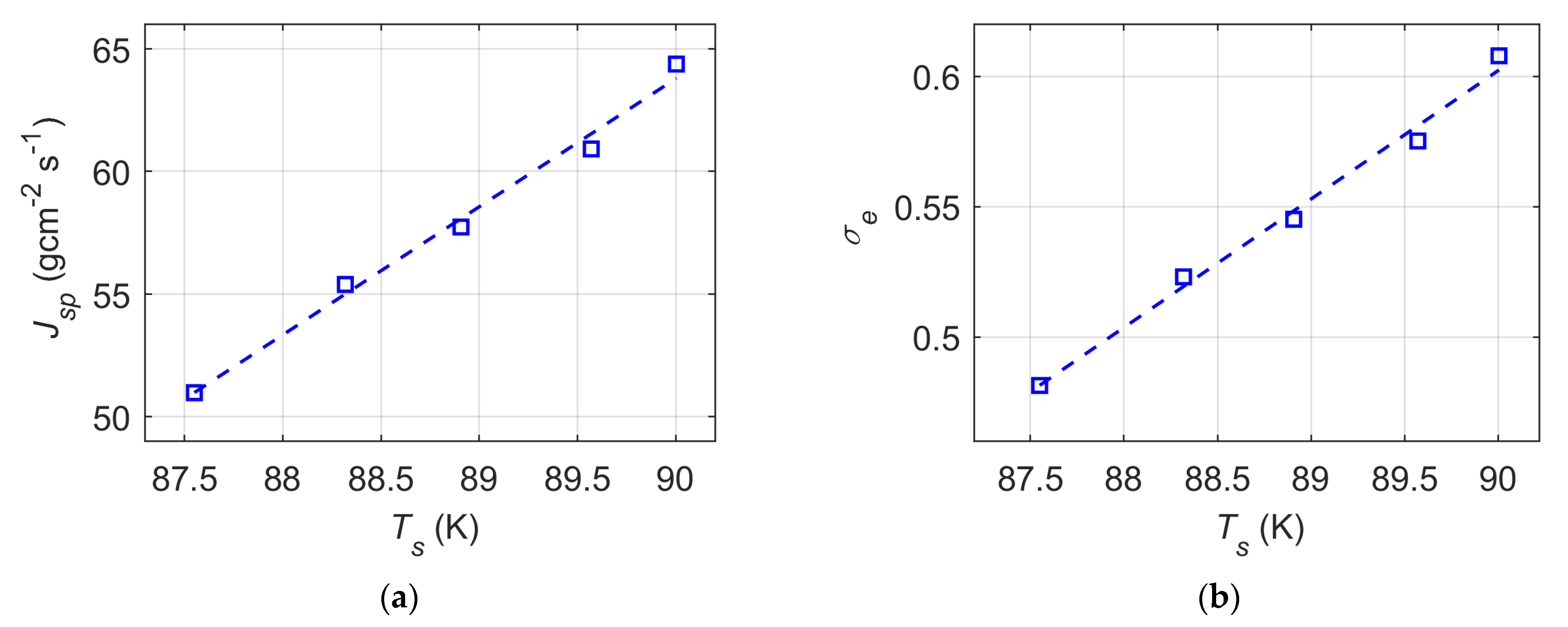
- The liquid surface temperature increases linearly with increasing normalized (or absolute) thermostat region boundary position values at the right-side interface; however, the surface temperature reaches the targeted simulation temperature of 90 K only when the thermostat boundary is placed beyond the liquid surface. Furthermore, the spontaneous evaporation rate is an increasing linear function of the liquid surface temperature in a temperature range of 87.55–90 K. In this narrow range, the spontaneous evaporation rate and corresponding evaporation coefficient estimation from MD simulations can vary up to 21%.
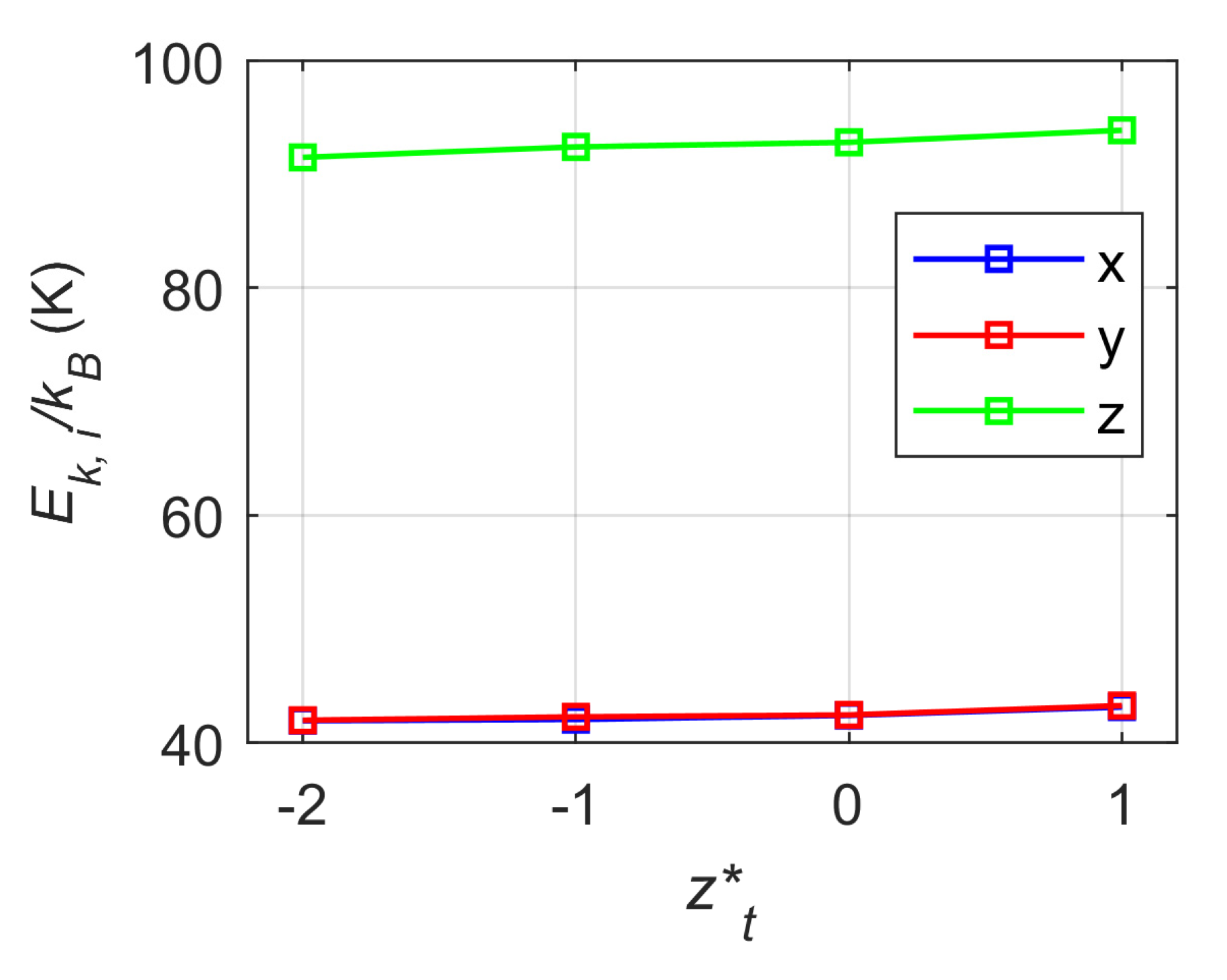
- The velocity distributions of evaporating argon atoms in x, y and z directions follow the Maxwellian distribution for all simulated cases, and the thermostat has little to no noticeable effect on the energy characteristics and velocity distributions of the evaporating argon atoms, even when the thermostat boundary is placed above the liquid surface.
- The proposed temperature control strategies can be used to obtain the evaporation/condensation coefficient values at desired liquid surface temperatures in practical applications.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tas, N.R.; Mela, P.; Kramer, T.; Berenschot, J.W.; Van Den Berg, A. Capillarity Induced Negative Pressure of Water Plugs in Nanochannels. Nano Lett. 2003, 3, 1537–1540. [Google Scholar] [CrossRef] [Green Version]
- Duan, C.; Karnik, R.; Lu, M.C.; Majumdar, A. Evaporation-induced cavitation in nanofluidic channels. Proc. Natl. Acad. Sci. USA 2012, 109, 3688–3693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajeel, R.K.; Salim, W.S.I.W.; Hasnan, K. Design characteristics of symmetrical semicircle-corrugated channel on heat transfer enhancement with nanofluid. Int. J. Mech. Sci. 2019, 151, 236–250. [Google Scholar] [CrossRef]
- Ajeel, R.K.; Zulkifli, R.; Sopian, K.; Fayyadh, S.N.; Fazlizan, A.; Ibrahim, A. Numerical investigation of binary hybrid nanofluid in new configurations for curved-corrugated channel by thermal-hydraulic performance method. Powder Technol. 2021, 385, 144–159. [Google Scholar] [CrossRef]
- Ajeel, R.K.; Sopian, K.; Zulkifli, R. A novel curved-corrugated channel model: Thermal-hydraulic performance and design parameters with nanofluid. Int. Commun. Heat Mass Transf. 2021, 120, 105037. [Google Scholar] [CrossRef]
- Baghbanzadeh, M.; Lan, C.Q.; Rana, D.; Matsuura, T. Membrane Distillation. J. Memb. Sci. 1997, 1, 419–455. [Google Scholar] [CrossRef]
- Lee, J.; Karnik, R. Desalination of water by vapor-phase transport through hydrophobic nanopores. J. Appl. Phys. 2010, 108, 044315. [Google Scholar] [CrossRef]
- Skarbalius, G.; Džiugys, A.; Misiulis, E.; Navakas, R.; Vilkinis, P.; Šereika, J.; Pedišius, N. Molecular dynamics study on water flow behaviour inside planar nanochannel using different temperature control strategies. Energies 2021, 14, 6843. [Google Scholar] [CrossRef]
- Cailliez, F.; Stirnemann, G.; Boutin, A.; Demachy, I.; Fuchs, A.H. Does water condense in hydrophobic cavities? A molecular simulation study of hydration in heterogeneous nanopores. J. Phys. Chem. C 2008, 112, 10435–10445. [Google Scholar] [CrossRef]
- Wu, X.; Yang, Z.; Duan, Y. Molecular dynamics simulation on evaporation of a suspending difluoromethane nanodroplet. Int. J. Heat Mass Transf. 2020, 158, 120024. [Google Scholar] [CrossRef]
- James, M.; Darwish, T.A.; Ciampi, S.; Sylvester, S.O.; Zhang, Z.; Ng, A.; Gooding, J.J.; Hanley, T.L. Nanoscale condensation of water on self-assembled monolayers. Soft Matter 2011, 7, 5309–5318. [Google Scholar] [CrossRef] [Green Version]
- Persad, A.H.; Ward, C.A. Expressions for the Evaporation and Condensation Coefficients in the Hertz-Knudsen Relation. Chem. Rev. 2016, 116, 7727–7767. [Google Scholar] [CrossRef] [PubMed]
- Hołyst, R.; Litniewski, M.; Jakubczyk, D. A molecular dynamics test of the Hertz-Knudsen equation for evaporating liquids. Soft Matter 2015, 11, 7201–7206. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Biben, T.; Keblinski, P. Molecular simulation of steady-state evaporation and condensation: Validity of the Schrage relationships. Int. J. Heat Mass Transf. 2017, 114, 105–114. [Google Scholar] [CrossRef]
- Barnes, G.T. The effects of monolayers on the evaporation of liquids. Adv. Colloid Interface Sci. 1986, 25, 89–200. [Google Scholar] [CrossRef]
- Mozurkewich, M. Aerosol growth and the condensation coefficient for water: A review. Aerosol Sci. Technol. 1986, 5, 223–236. [Google Scholar] [CrossRef]
- Marek, R.; Straub, J. Analysis of the evaporation coefficient and the condensation coefficient of water. Int. J. Heat Mass Transf. 2001, 44, 39–53. [Google Scholar] [CrossRef]
- Davis, E.J. A history and state-of-the-art of accommodation coefficients. Atmos. Res. 2006, 82, 561–578. [Google Scholar] [CrossRef]
- Badam, V.K.; Kumar, V.; Durst, F.; Danov, K. Experimental and theoretical investigations on interfacial temperature jumps during evaporation. Exp. Therm. Fluid Sci. 2007, 32, 276–292. [Google Scholar] [CrossRef]
- Gatapova, E.Y.; Graur, I.A.; Kabov, O.A.; Aniskin, V.M.; Filipenko, M.A.; Sharipov, F.; Tadrist, L. The temperature jump at water—Air interface during evaporation. Int. J. Heat Mass Transf. 2017, 104, 800–812. [Google Scholar] [CrossRef]
- Rokoni, A.; Sun, Y. Probing the temperature profile across a liquid-vapor interface upon phase change. J. Chem. Phys. 2020, 153, 144706. [Google Scholar] [CrossRef] [PubMed]
- Bird, E.; Liang, Z. Transport phenomena in the Knudsen layer near an evaporating surface. Phys. Rev. E 2019, 100, 43108. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Lechman, J.B.; Plimpton, S.J.; Grest, G.S. Evaporation of Lennard-Jones fluids. J. Chem. Phys. 2011, 134, 224704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishiyama, T.; Yano, T.; Fujikawa, S. Molecular dynamics study of kinetic boundary condition at an interface between argon vapor and its condensed phase. Phys. Fluids 2004, 16, 2899. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.F.; Sazhin, S.S.; Cao, B.Y. Molecular dynamics study of the processes in the vicinity of the n-dodecane vapour/liquid interface. Phys. Fluids 2011, 23, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ishiyama, T.; Yano, T.; Fujikawa, S. Molecular dynamics study of kinetic boundary condition at an interface between a polyatomic vapor and its condensed phase. Phys. Fluids 2004, 16, 4713–4727. [Google Scholar] [CrossRef]
- Ishiyama, T.; Fujikawa, S.; Kurz, T.; Lauterborn, W. Nonequilibrium kinetic boundary condition at the vapor-liquid interface of argon. Phys. Rev. E—Stat. Nonlinear Soft Matter Phys. 2013, 88, 042406. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Hori, K.; Yaguchi, H.; Watanabe, M. Molecular dynamics simulation on evaporation molecules in a vapor-liquid equilibrium state. Proc. 29th Int. Symp. Rarefied Gas Dyn. 2014, 1628, 404–410. [Google Scholar] [CrossRef]
- Kobayashi, K.; Hori, K.; Kon, M.; Sasaki, K.; Watanabe, M. Molecular dynamics study on evaporation and reflection of monatomic molecules to construct kinetic boundary condition in vapor—Liquid equilibria. Heat Mass Transf. 2015, 52, 1851–1859. [Google Scholar] [CrossRef] [Green Version]
- Bird, E.; Gutierrez Plascencia, J.; Keblinski, P.; Liang, Z. Molecular simulation of steady-state evaporation and condensation of water in air. Int. J. Heat Mass Transf. 2022, 184, 122285. [Google Scholar] [CrossRef]
- Yang, T.H.; Pan, C. Molecular dynamics simulation of a thin water layer evaporation and evaporation coefficient. Int. J. Heat Mass Transf. 2005, 48, 3516–3526. [Google Scholar] [CrossRef]
- Skarbalius, G.; Džiugys, A.; Misiulis, E.; Navakas, R. Molecular dynamics study on water evaporation/condensation parameters. Microfluid. Nanofluidics 2021, 25, 81. [Google Scholar] [CrossRef]
- Zhakhovsky, V.V.; Kryukov, A.P.; Levashov, V.Y.; Shishkova, I.N.; Anisimov, S.I. Mass and heat transfer between evaporation and condensation surfaces: Atomistic simulation and solution of Boltzmann kinetic equation. Proc. Natl. Acad. Sci. USA 2019, 116, 18209–18217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sam, A.; Kannam, S.K.; Hartkamp, R.; Sathian, S.P. Water flow in carbon nanotubes: The effect of tube flexibility and thermostat. J. Chem. Phys. 2017, 146, 234701. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.P.; Aktulga, H.M.; Berger, R.; Bolintineanu, D.S.; Brown, W.M.; Crozier, P.S.; in ’t Veld, P.J.; Kohlmeyer, A.; Moore, S.G.; Nguyen, T.D.; et al. LAMMPS—A flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 2022, 271, 108171. [Google Scholar] [CrossRef]
- Liang, Z.; Chandra, A.; Bird, E.; Keblinski, P. A molecular dynamics study of transient evaporation and condensation. Int. J. Heat Mass Transf. 2020, 149, 119152. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| −36.63 | 36.64 | 4.17 | 4.18 | 9.11 |
| Left vapor boundary | 3.0 | −64.0 |
| Left liquid boundary | −0.9 | −27.5 |
| Right liquid boundary | −0.9 | 27.5 |
| Right vapor boundary | 3.0 | 64.0 |
| Left vapor boundary | 3.0 | −64.0 |
| Reflection boundary | 2.58 | −60.2 |
| Limit NVE boundary | 2.16 | −56.3 |
| Left liquid boundary | −0.9 | −27.5 |
| Right liquid boundary | −0.9 | 27.5 |
| Right vapor boundary | 3.0 | 64.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skarbalius, G.; Džiugys, A.; Misiulis, E.; Navakas, R. The Impact of the Temperature Control Strategy in Steady-State Virtual Vacuum Simulation on the Spontaneous Evaporation Rate and Corresponding Evaporation Coefficient. Appl. Sci. 2023, 13, 256. https://doi.org/10.3390/app13010256
Skarbalius G, Džiugys A, Misiulis E, Navakas R. The Impact of the Temperature Control Strategy in Steady-State Virtual Vacuum Simulation on the Spontaneous Evaporation Rate and Corresponding Evaporation Coefficient. Applied Sciences. 2023; 13(1):256. https://doi.org/10.3390/app13010256
Chicago/Turabian StyleSkarbalius, Gediminas, Algis Džiugys, Edgaras Misiulis, and Robertas Navakas. 2023. "The Impact of the Temperature Control Strategy in Steady-State Virtual Vacuum Simulation on the Spontaneous Evaporation Rate and Corresponding Evaporation Coefficient" Applied Sciences 13, no. 1: 256. https://doi.org/10.3390/app13010256