
Pharmaceuticals in Coastal Waters: An UHPLC-TOF-MS Multi-Residue Approach

 ,
,  ,
,  , ,
, ,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Standards and Reagents
2.2. Sample Treatment
2.3. Instrumentation
2.4. Validation Procedure
3. Results and Discussion
3.1. Method Development
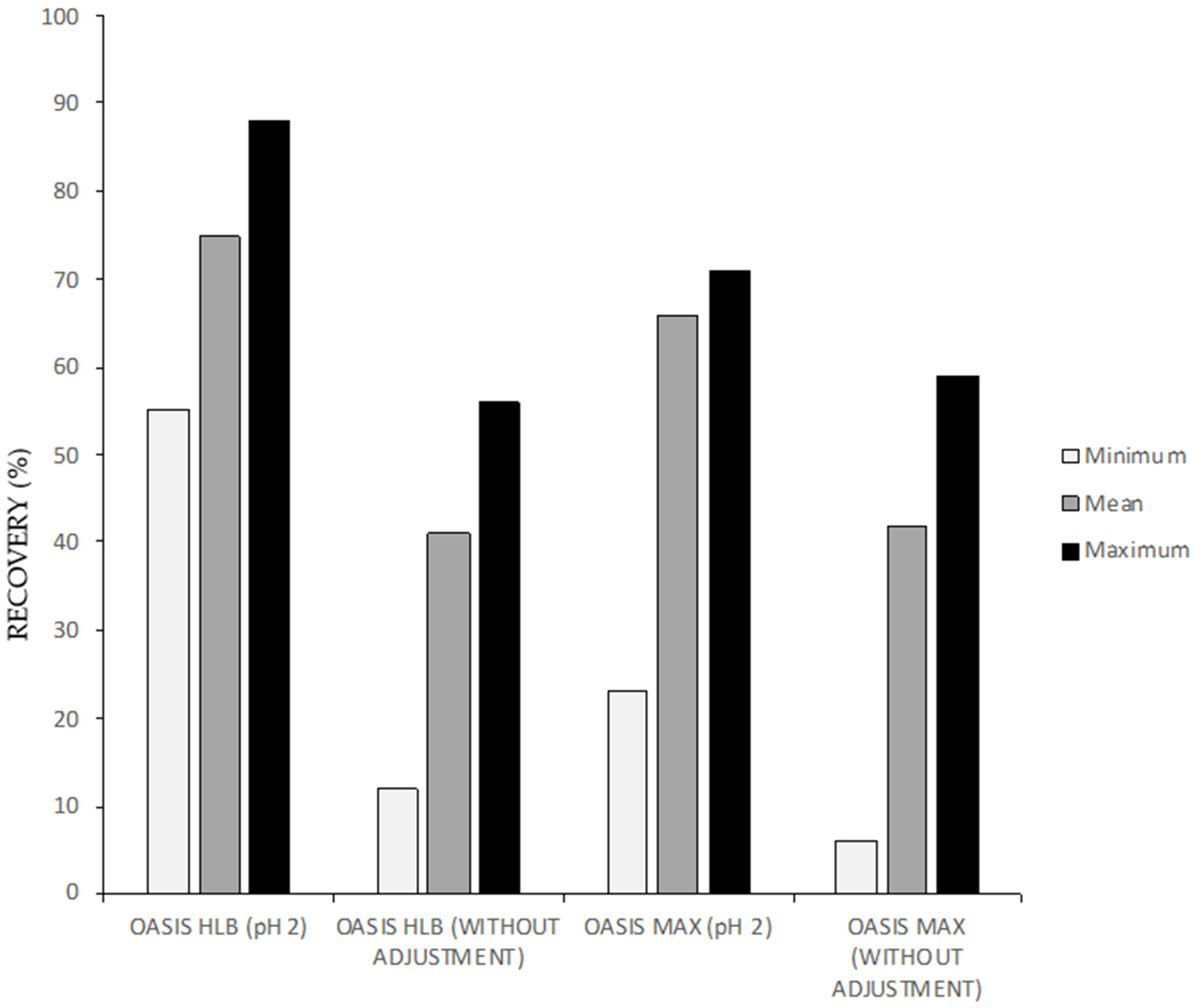
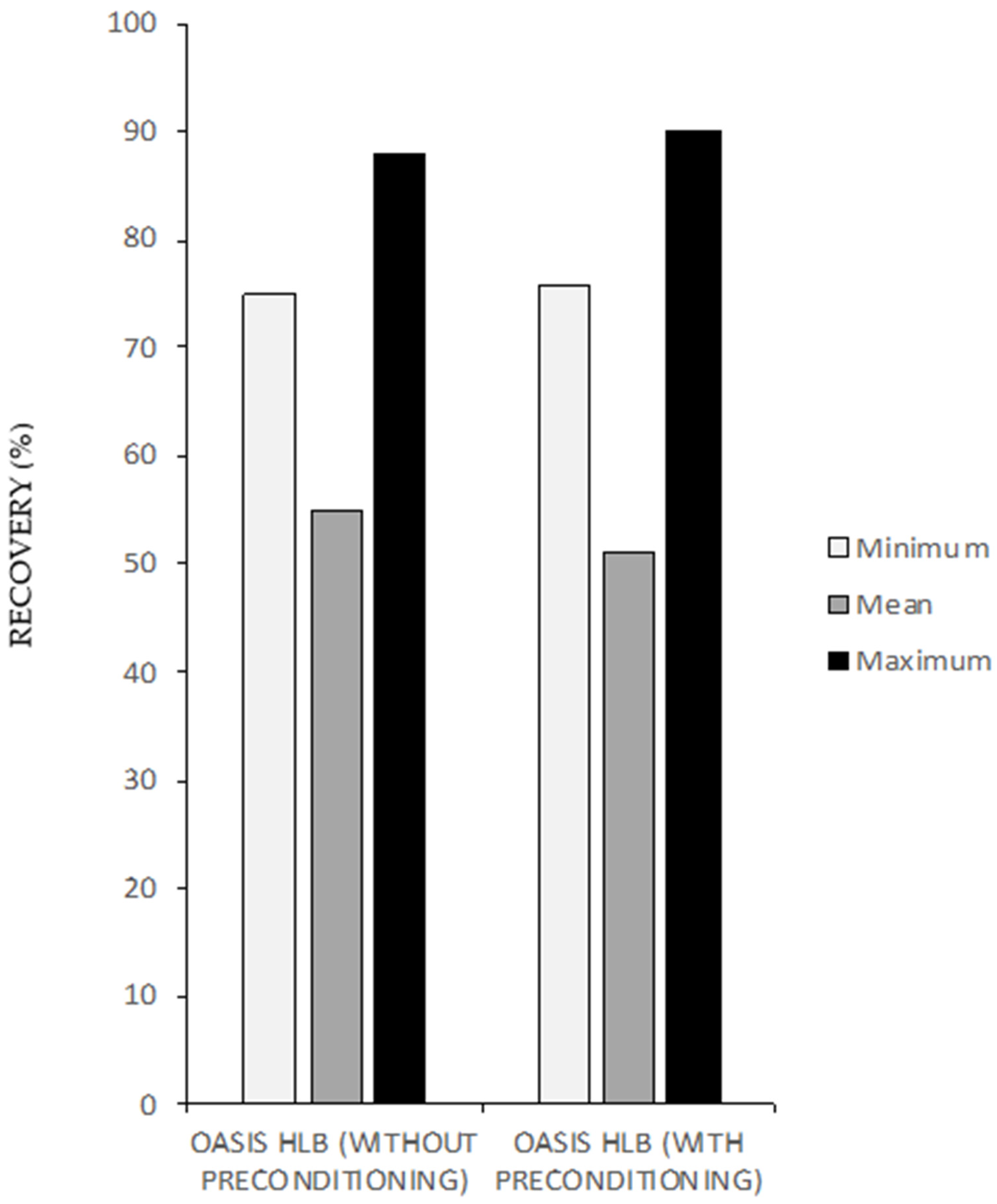
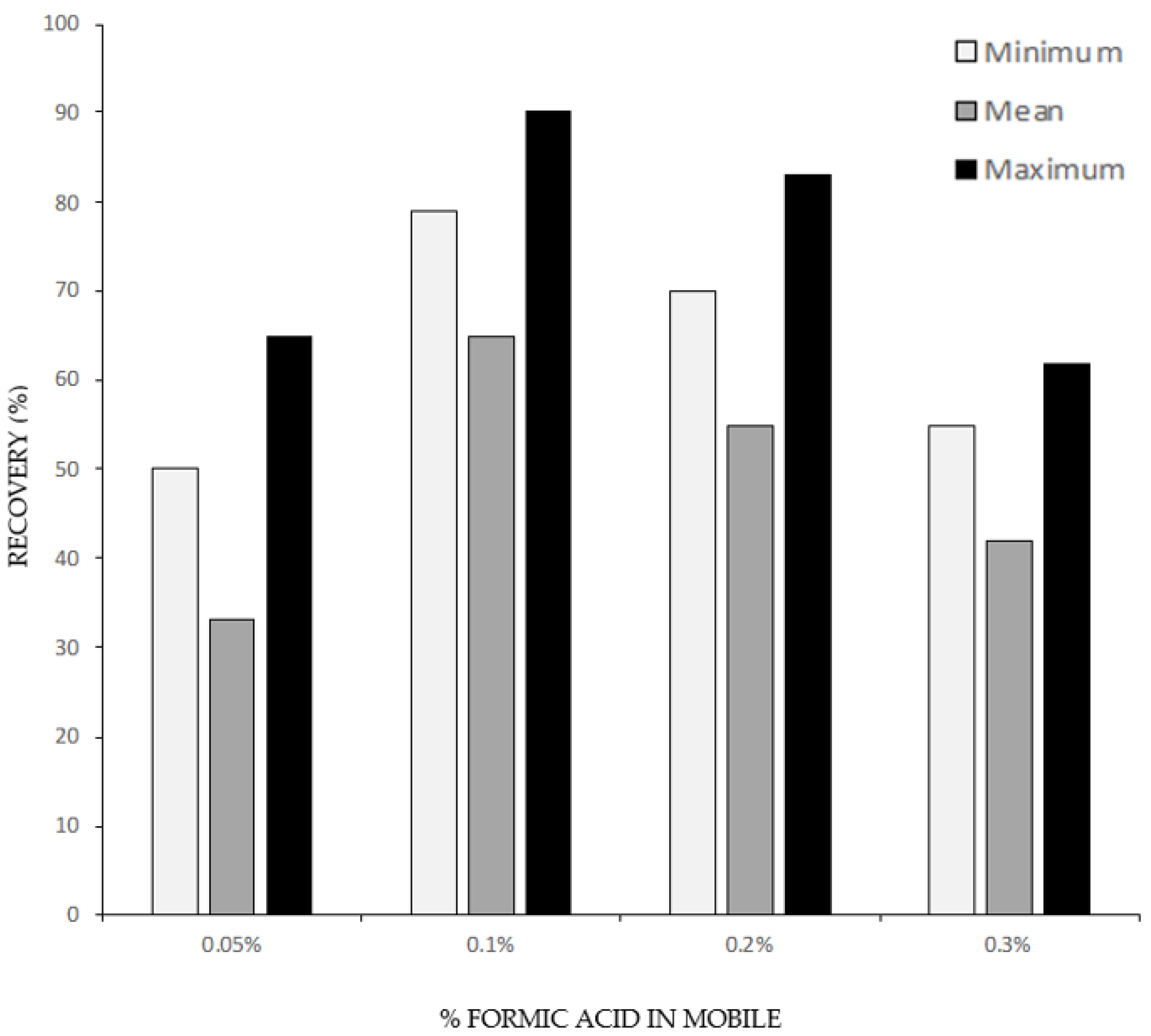
3.1.1. Solid Phase Extraction
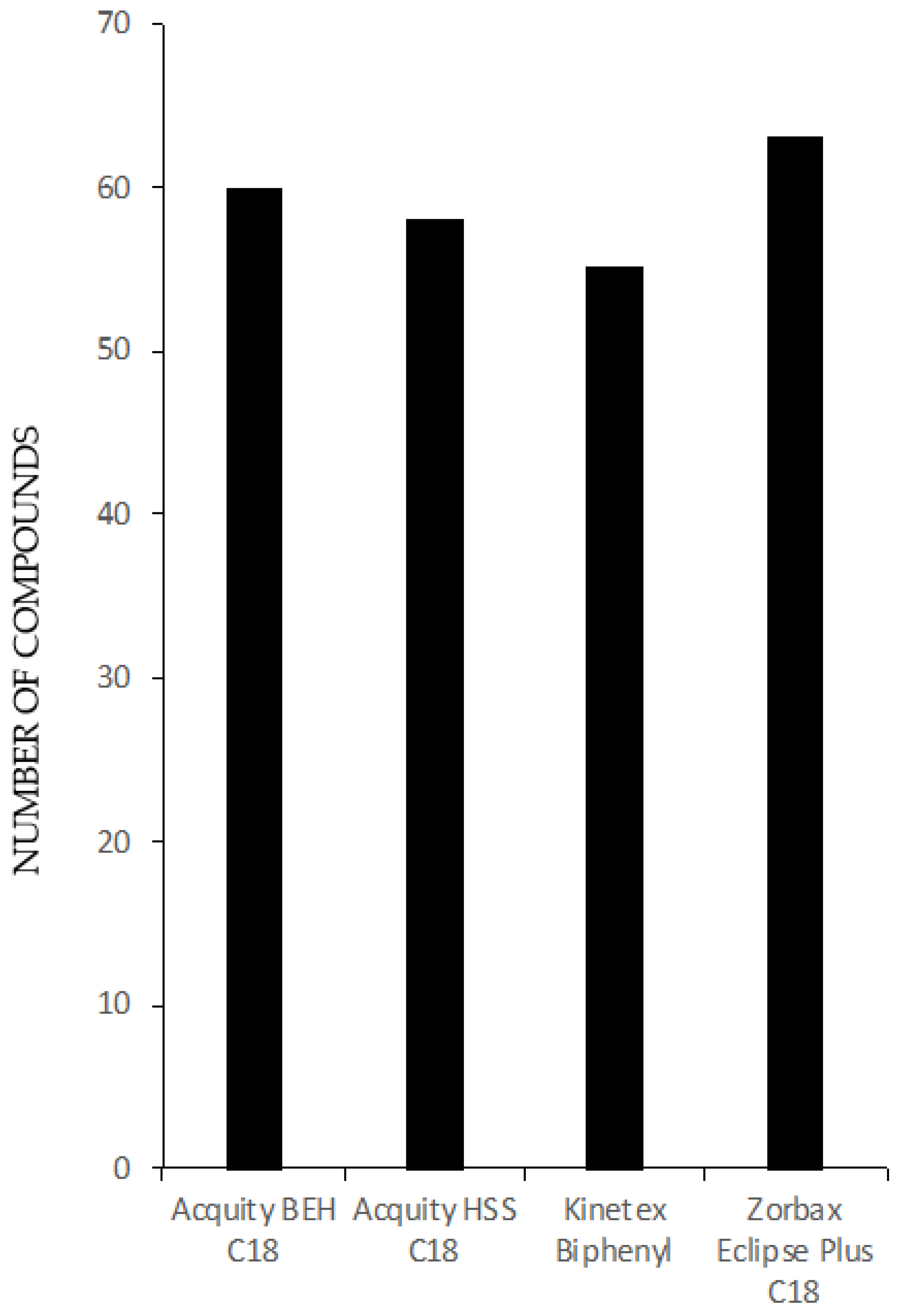
3.1.2. UHPLC-TOF-MS Performance
3.2. Validation
3.3. Application to Field Samples
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iglesias-Campos, A.; Meiner, A.; Bowen, K.; Onwona Ansong, J. Chapter 3—Coastal Population and Land Use Changes in Juan; Baztan, J., Chouinard, O., Jorgensen, B., Tett, P., Vanderlinden, J.-P., Vasseur, L., Eds.; Europe: Challenges for a Sustainable Future in Coastal Zones; Elsevier: Amsterdam, The Netherlands, 2015; pp. 29–49. [Google Scholar] [CrossRef]
- Nunes, M.; Leston, S. Coastal Pollution: An Overview. In Life Below Water. Encyclopedia of the UN Sustainable Development Goals; Leal Filho, W., Azul, A.M., Brandli, L., Lange Salvia, A., Wall, T., Eds.; Springer: Cham, Switzerland, 2020. [Google Scholar] [CrossRef]
- Riera, R.; Menci, C.; Sanabria-Fernández, J.A.; Becerro, M.A. Do recreational activities affect coastal biodiversity? Estuar. Coast. Shelf Sci. 2016, 178, 129–136. [Google Scholar] [CrossRef]
- United Nations. The Second World Ocean Assessment II; Cambridge University Press: Cambridge, UK, 2021.
- Branchet, P.; Arpin-Pont, L.; Piram, A.; Boissery, P.; Wong-Wah-Chung, P.; Doumenq, P. Pharmaceuticals in the marine environment: What are the present challenges in their monitoring? Sci. Total Environ. 2021, 766, 142644. [Google Scholar] [CrossRef] [PubMed]
- Desbiolles, F.; Malleret, L.; Tiliacos, C.; Wong-Wah-Chung, P.; Laffont-Schwob, P. Occurrence and ecotoxicological assessment of pharmaceuticals: Is there a risk for the Mediterranean aquatic environment? Sci. Total Environ. 2018, 639, 1334–1348. [Google Scholar] [CrossRef] [PubMed]
- Leston, S.; Freitas, A.; Rosa, J.; Barbosa, J.; Lemos, M.F.L.; Pardal, M.A.; Ramos, F. A multiresidue approach for the simultaneous quantification of antibiotics in macroalgae by ultra-high performance liquid chromatography–tandem mass spectrometry. J. Chromatogr. B 2016, 1033, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Shi, W.; Li, H.; Xu, N.; Zhang, R.; Chen, X.; Sun, W.; Wen, D.; He, S.; Pan, J.; et al. Antibiotics in water and sediments of rivers and coastal area of Zhuhai City, Pearl River estuary, South China. Sci. Total Environ. 2018, 636, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Kümmerer, K. Antibiotics in the aquatic environment—A review—Part I. Chemosphere 2009, 75, 417–434. [Google Scholar] [CrossRef] [PubMed]
- Loos, R.; Marinov, D.; Sanseverino, I.; Napiersk, D.; Lettieri, T. Review of the 1st Watch List under the Water Framework Directive and Recommendations for the 2nd Watch List, EUR 29173 EN; JRC; Publications Office of the European Union: Luxembourg, 2018; ISBN 978-92-79-81839-4. [Google Scholar] [CrossRef]
- OSPAR Commission. JAMP Guidelines for Monitoring of Contaminants in Seawater. 2013. Available online: https://mcc.jrc.ec.europa.eu/documents/OSPAR/Guidelines_forMonitoring_of_ContaminantsSeawater.pdf (accessed on 3 February 2023).
- INFARMED. Medicine and Healthcare Products Statistics. 2017. Available online: https://www.infarmed.pt/web/infarmed/entidades/medicamentos-uso-humano/monitorizacao-mercado/estatistica-anual/relatorios-anuais (accessed on 3 February 2023).
- Sousa, M.A.; Gonçalves, C.; Cunha, E.; Hajšlová, J.; Alpendurada, M.F. Cleanup strategies and advantages in the determination of several therapeutic classes of pharmaceuticals in wastewater samples by SPE–LC–MS/MS. Anal. Bioanal. Chem. 2011, 399, 807–822. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.M.P.T.; Silva, L.J.G.; Meisel, L.M.; Lino, C.M.; Pena, A. Environmental impact of pharmaceuticals from Portuguese wastewaters: Geographical and seasonal occurrence, removal and risk assessment. Environ. Res. 2015, 136, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Commission Implementing Decision (EU) 2020/1161. Official Journal of the European Union, L 257/32. 2020. Available online: http://data.europa.eu/eli/dec_impl/2020/1161/oj (accessed on 3 February 2023).
- Commission Implementing Regulation (EU) Official Journal of the European Union, L 180/84. 2021. Available online: http://data.europa.eu/eli/reg_impl/2021/808/oj (accessed on 3 February 2023).
- Magalhães, D.; Freitas, A.; Vila Pouca, A.S.; Barbosa, J.; Ramos, F. The use of ultra-high-pressure-liquid-chromatography tandem time-of-flight mass spectrometry as a confirmatory method in drug residue analysis: Application to the determination of antibiotics in piglet liver. J. Chromatogr. B 2020, 1153, 122264. [Google Scholar] [CrossRef] [PubMed]
- Reis-Santos, P.; Pais, M.; Duarte, B.; Caçador, I.; Freitas, A.; Vila Pouca, A.S.; Barbosa, J.; Leston, S.; Rosa, J.; Ramos, F.; et al. Screening of human and veterinary pharmaceuticals in estuarine waters: A baseline assessment for the Tejo estuary. Mar. Pollut. Bull. 2018, 135, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, V.F.; Duarte, I.A.; Duarte, B.; Freitas, A.; Pouca AS, V.; Barbosa, J.; Gillanders, B.M.; Reis-Santos, P. Environmental risk assessment and bioaccumulation of pharmaceuticals in a large urbanized estuary. Sci. Total Environ. 2021, 783, 147021. [Google Scholar] [CrossRef] [PubMed]
- Moyo, B.; Tawanda Tavengwa, N. Modern Extraction and Cleanup Methods of Veterinary Drug Residues in Food Samples of Animal Origin. Recent Adv. Anal. Chem. 2019, 1, 21. [Google Scholar] [CrossRef]
- Nurmi, J.; Pellinen, J. Multiresidue method for the analysis of emerging contaminants in wastewater by ultra performance liquid chromatography–time-of-flight mass spectrometry. J. Chromatogr. A 2011, 1218, 6712–6719. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, M.; Gros, M.; Barcelo, D. Multi-residue analysis of pharmaceuticals in wastewater by ultra-performance liquid chromatography–quadrupole–time-of-flight mass spectrometry. J. Chromatogr. A 2006, 1124, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Kiontke, A.; Oliveira-Birkmeier, A.; Opitz, A.; Birkemeyer, C. Electrospray Ionization Efficiency Is Dependent on Different Molecular Descriptors with Respect to Solvent pH and Instrumental Configuration. PLoS ONE 2016, 11, e0167502. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Molecular Formula | LogP | Molecular Weight | [M + H]+ | ΔM (ppm) | LoD (ng/L) | LoQ (ng/L) | Repeatability (%) | Reproducibility (%) | Recovery (%) | RT (min) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Analgesic | |||||||||||
| Acetaminophen | C8H9NO2 | 0.51 | 151.0633 | 152.0706 | −0.8 | 1.89 | 6.30 | 3.0% | 4.5% | 99.8% | 2.21 |
| Antibiotics | |||||||||||
| Amoxicillin | C16H19N3O5S | 0.75 | 365.1045 | 366.1118 | 2.3 | 0.11 | 0.38 | 16.2% | 20.9% | 107.2% | 6.91 |
| Azithromycin | C38H72N2O12 | 3.03 | 748.5085 | 749.5158 | −0.9 | 0.01 | 0.04 | 11.5% | 14.4% | 96.0% | 5.04 |
| Benzylpenicillin | C16H18N2O4S | 1.83 | 334.0987 | 335.1060 | 0.1 | 0.75 | 2.52 | 3.1% | 4.7% | 88.7% | 4.49 |
| Ceftiofur | C19H17N5O7S3 | 1.22 | 523.0290 | 524.0363 | 1.3 | 0.03 | 0.09 | 10.5% | 16.8% | 94.7% | 5.85 |
| Cephalexin | C16H17N3O4S | 0.55 | 347.0939 | 348.1013 | 0.5 | 0.04 | 0.10 | 16.6% | 18.5% | 98.6% | 4.85 |
| Chlortetracycline | C22H23ClN2O8 | −0.13 | 478.1142 | 479.1216 | 0.7 | 1.73 | 5.77 | 2.2% | 3.2% | 99.5% | 4.78 |
| Cinoxacin | C12H10N2O5 | 1.25 | 262.0589 | 263.0663 | −0.8 | 0.03 | 0.09 | 14.9% | 16.9% | 92.5% | 5.25 |
| Ciprofloxacin | C17H18FN3O3 | −0.57 | 331.1332 | 332.1405 | 0.6 | 3.47 | 11.56 | 3.2% | 4.7% | 110.5% | 4.47 |
| Danofloxacin | C19H20FN3O3 | 0.33 | 357.1488 | 358.1562 | 0.5 | 3.25 | 10.82 | 6.5% | 9.7% | 110.1% | 4.59 |
| Doxycyclin | C22H24N2O8 | −0.72 | 444.1532 | 445.1605 | −0.6 | 0.28 | 0.94 | 6.7% | 9.5% | 101.5% | 5.23 |
| Enoxacin | C15H17FN4O3 | −0.97 | 320.1284 | 321.1358 | 0.9 | 3.33 | 11.09 | 3.9% | 5.8% | 109.5% | 4.33 |
| Enrofloxacin | C19H22FN3O3 | 0.58 | 359.1645 | 360.1718 | 0.6 | 2.27 | 7.58 | 2.5% | 3.8% | 101.4% | 4.66 |
| Epi-Chlortetracycline | C22H23ClN2O8 | −0.13 | 478.1142 | 479.1216 | 0.6 | 1.00 | 3.33 | 2.2% | 3.3% | 99.0% | 4.54 |
| epi-Tetracycline | C22H24N2O8 | −0.56 | 444.1532 | 445.1605 | 1.0 | 0.40 | 1.32 | 6.5% | 7.2% | 104.7% | 4.27 |
| Flumequine | C14H12FNO3 | 1.62 | 261.0801 | 262.0874 | −0.3 | 0.01 | 0.04 | 8.2% | 10.1% | 80.6% | 6.18 |
| Marbofloxacin | C17H19FN4O4 | −0.53 | 362.1390 | 363.1463 | 0.6 | 2.51 | 8.35 | 3.1% | 4.6% | 108.6% | 4.29 |
| Nalidixic acid | C12H12N2O3 | 0.95 | 232.0847 | 233.0921 | −0.9 | 0.92 | 3.07 | 4.2% | 6.3% | 106.0% | 6.07 |
| Norfloxacin | C16H18FN3O3 | −0.47 | 319.1332 | 320.1405 | −0.4 | 1.81 | 6.04 | 9.0% | 10.2% | 109.6% | 4.41 |
| Ofloxacin | C18H20FN3O4 | −0.02 | 361.1437 | 362.1511 | 0.6 | 0.99 | 3.31 | 1.4% | 2.6% | 91.5% | 4.43 |
| Oxolinic acid | C13H11NO5 | 0.86 | 261.0637 | 262.0710 | 0.6 | 3.08 | 10.26 | 3.4% | 7.1% | 92.3% | 5.50 |
| Oxytetracycline | C22H24N2O9 | −0.99 | 460.1481 | 461.1555 | 0.3 | 0.24 | 0.79 | 7.6% | 11.4% | 104.3% | 4.40 |
| Spiramycin | C43H74N2O14 | 2.99 | 842.5140 | 843.5213 | −0.2 | 0.01 | 0.04 | 5.6% | 7.1% | 87.7% | 5.02 |
| Sulfachloropyridazine | C10H9ClN4O2S | 0.97 | 284.0134 | 285.0208 | −1.0 | 0.43 | 1.43 | 6.0% | 7.0% | 103.9% | 4.95 |
| Sulfadiazine | C10H10N4O2S | 0.25 | 250.0524 | 251.0597 | −1.1 | 0.01 | 0.02 | 5.5% | 8.2% | 107.5% | 3.81 |
| Sulfadimethoxine | C12H14N4O4S | 1.08 | 310.0735 | 311.0809 | −0.9 | 0.30 | 0.98 | 2.7% | 4.1% | 100.3% | 5.70 |
| Sulfadimidin | C12H14N4O2S | 0.43 | 278.0837 | 279.091 | −0.9 | 0.14 | 0.47 | 1.9% | 2.5% | 100.9% | 4.51 |
| Sulfadoxine | C12H14N4O4S | 0.72 | 310.0735 | 311.0809 | −0.7 | 0.03 | 0.10 | 6.6% | 7.7% | 98.3% | 4.94 |
| Sulfamethizole | C9H10N4O2S2 | 0.53 | 270.0245 | 271.0318 | −1.3 | 0.32 | 1.06 | 12.7% | 15.2% | 100.0% | 4.52 |
| Sulfamethoxazole | C10H11N3O3S | 0.79 | 253.0521 | 254.0594 | −0.7 | 0.03 | 0.09 | 2.5% | 3.7% | 99.5% | 5.12 |
| Sulfapyridine | C11H11N3O2S | 0.84 | 249.0572 | 250.0645 | −0.7 | 0.46 | 1.52 | 3.3% | 3.3% | 104.4% | 3.80 |
| Sulfaquinoxaline | C14H12N4O2S | 1.24 | 300.0681 | 301.0754 | −0.7 | 0.03 | 0.09 | 4.5% | 6.7% | 88.3% | 5.71 |
| Sulfathiazole | C9H9N3O2S2 | 0.88 | 255.0136 | 256.0209 | −1.3 | 0.56 | 1.87 | 6.6% | 10.6% | 108.6% | 3.65 |
| Sulfisomidine | C12H14N4O2S | 0.84 | 278.0837 | 279.091 | −0.2 | 0.10 | 0.32 | 9.8% | 14.6% | 87.0% | 3.42 |
| Sulfisoxazole | C11H13N3O3S | 1.14 | 267.0677 | 268.075 | 0.1 | 0.02 | 0.07 | 9.8% | 13.5% | 101.1% | 5.04 |
| Tetracycline | C22H24N2O8 | −0.56 | 444.1532 | 445.1605 | 0.2 | 0.48 | 1.61 | 5.5% | 8.2% | 112.6% | 4.57 |
| Tilmicosin | C46H80N2O13 | 3.34 | 868.5660 | 869.5733 | −0.9 | 0.01 | 0.05 | 3.7% | 4.5% | 95.9% | 5.40 |
| Trimethoprim | C14H18N4O3 | 1.26 | 290.1378 | 291.1452 | 0.3 | 0.80 | 2.68 | 4.3% | 6.4% | 109.5% | 4.24 |
| Tylosin A | C46H77NO17 | 1.46 | 915.5191 | 916.5264 | −0.9 | 0.36 | 1.21 | 4.6% | 6.1% | 86.7% | 5.90 |
| Anticonvulsants | |||||||||||
| Carbamazepine | C15H12N2O | 2.10 | 237.1022 | 237.1022 | −0.8 | 0.01 | 0.05 | 12.5% | 18.7% | 102.9% | 6.08 |
| Gabapentin | C9H17NO2 | −1.90 | 172.1332 | 172.1332 | −0.5 | 0.81 | 2.69 | 4.1% | 6.1% | 80.6% | 3.61 |
| Topiramate | C12H21NO8S | 1.29 | 340.1061 | 340.1061 | 0.6 | 0.03 | 0.11 | 2.9% | 3.7% | 104.7% | 5.82 |
| Antidepressants | |||||||||||
| Alpha-Hydroxyalprazolam | C17H13ClN4O | 1.53 | 324.0777 | 325.0851 | −0.3 | 0.02 | 0.08 | 5.6% | 8.3% | 84.9% | 6.12 |
| Fluoxetine | C17H18F3NO | 4.09 | 309.1340 | 310.1413 | −0.2 | 0.01 | 0.03 | 5.2% | 7.8% | 109.3% | 4.88 |
| Lorazepam | C15H10Cl2N2O2 | 2.98 | 320.0119 | 321.0192 | 3.1 | 2.98 | 9.94 | 7.6% | 8.7% | 108.9% | 4.53 |
| Sertraline | C17H17Cl2N | 5.06 | 305.0738 | 306.0811 | 0.4 | 0.03 | 0.09 | 3.2% | 4.0% | 109.6% | 6.13 |
| Venlafaxine | C17H27NO2 | 2.69 | 277.2041 | 278.2115 | −0.9 | 0.02 | 0.08 | 6.0% | 7.3% | 99.6% | 5.24 |
| Antihypertensives | |||||||||||
| Furosemide | C12H11ClN2O5S | 2.71 | 331.015 | 331.015 | −1.0 | 0.62 | 2.07 | 7.7% | 11.6% | 109.3% | 6.04 |
| Indapamide | C16H16ClN3O3S | 2.52 | 366.0674 | 366.0674 | −0.3 | 0.03 | 0.09 | 3.6% | 4.0% | 100.4% | 6.27 |
| Irbesartan | C25H28N6O | 4.51 | 429.2397 | 429.2397 | −0.7 | 0.03 | 0.17 | 10.5% | 15.7% | 96.0% | 6.31 |
| Losartan | C22H23ClN6O | 4.50 | 423.1695 | 423.1695 | −0.5 | 0.05 | 0.09 | 12.3% | 18.4% | 107.1% | 5.77 |
| β-Blockers | |||||||||||
| Atenolol | C14H22N2O3 | 0.57 | 266.1630 | 267.1703 | −0.7 | 0.01 | 0.03 | 3.0% | 4.2% | 104.4% | 3.47 |
| Bisoprolol | C18H31NO4 | 2.30 | 325.2253 | 326.2326 | −1.0 | 0.14 | 0.46 | 8.3% | 9.1% | 94.7% | 5.29 |
| Carvedilol | C24H26N2O4 | 3.05 | 406.1892 | 407.1965 | −0.9 | 0.83 | 2.76 | 13.4% | 19.7% | 108.7% | 5.87 |
| Propranolol | C16H21NO2 | 3.03 | 259.1572 | 260.1645 | −0.6 | 0.06 | 0.21 | 4.5% | 6.5% | 100.9% | 4.39 |
| Lipid regulators | |||||||||||
| Atorvastatin | C33H35FN2O5 | 4.24 | 559.2603 | 559.2603 | 0.6 | 8.92 | 29.73 | 7.9% | 10.2% | 88.6% | 8.46 |
| Bezafibrate | C19H20ClNO4 | 3.97 | 362.1154 | 362.1154 | −0.8 | 0.07 | 0.24 | 7.4% | 8.6% | 85.7% | 6.91 |
| Fenofibrate | C20H21ClO4 | 4.86 | 361.1201 | 361.1201 | 0.7 | 0.02 | 0.06 | 17.2% | 18.1% | 103.5% | 9.41 |
| Gemfibrozil | C15H22O3 | 3.61 | 251.1642 | 251.1642 | −0.8 | 0.21 | 0.71 | 4.5% | 6.8% | 110.4% | 4.99 |
| Simvastatin | C25H38O5 | 4.51 | 419.2792 | 419.2792 | −1.6 | 2.80 | 9.34 | 11.7% | 12.2% | 110.3% | 6.20 |
| NSAID’s | |||||||||||
| Diclofenac | C14H11Cl2NO2 | 4.98 | 295.0166 | 296.024 | −0.5 | 0.02 | 0.05 | 5.9% | 8.8% | 106.0% | 7.70 |
| Ibuprofen | C13H18O2 | 3.50 | 206.1306 | 207.138 | −0.4 | 2.72 | 9.08 | 3.5% | 5.3% | 108.9% | 6.22 |
| Nimesulide | C13H12N2O5S | 2.56 | 308.0466 | 309.054 | 0.3 | 0.03 | 0.08 | 7.4% | 11.1% | 104.5% | 7.23 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leston, S.; Freitas, A.; Rosa, J.; Vila Pouca, A.S.; Barbosa, J.; Reis-Santos, P.; Fonseca, V.F.; Pardal, M.A.; Ramos, F. Pharmaceuticals in Coastal Waters: An UHPLC-TOF-MS Multi-Residue Approach. Appl. Sci. 2023, 13, 5975. https://doi.org/10.3390/app13105975
Leston S, Freitas A, Rosa J, Vila Pouca AS, Barbosa J, Reis-Santos P, Fonseca VF, Pardal MA, Ramos F. Pharmaceuticals in Coastal Waters: An UHPLC-TOF-MS Multi-Residue Approach. Applied Sciences. 2023; 13(10):5975. https://doi.org/10.3390/app13105975
Chicago/Turabian StyleLeston, Sara, Andreia Freitas, João Rosa, Ana Sofia Vila Pouca, Jorge Barbosa, Patrick Reis-Santos, Vanessa F. Fonseca, Miguel A. Pardal, and Fernando Ramos. 2023. "Pharmaceuticals in Coastal Waters: An UHPLC-TOF-MS Multi-Residue Approach" Applied Sciences 13, no. 10: 5975. https://doi.org/10.3390/app13105975
APA StyleLeston, S., Freitas, A., Rosa, J., Vila Pouca, A. S., Barbosa, J., Reis-Santos, P., Fonseca, V. F., Pardal, M. A., & Ramos, F. (2023). Pharmaceuticals in Coastal Waters: An UHPLC-TOF-MS Multi-Residue Approach. Applied Sciences, 13(10), 5975. https://doi.org/10.3390/app13105975