
Non-Enzymatic Formation of N-acetylated Amino Acid Conjugates in Urine

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagent Preparation and Chemicals
2.2. Sample Availability
2.3. Instrumentation
2.4. Methods
2.4.1. Organic Acid Analysis
2.4.2. N-AAA Conjugates Hydrolysis
2.4.3. Non-Enzymatic In Vitro Synthesis of N-AAA Conjugates
2.4.4. Molecular Modelling
3. Results
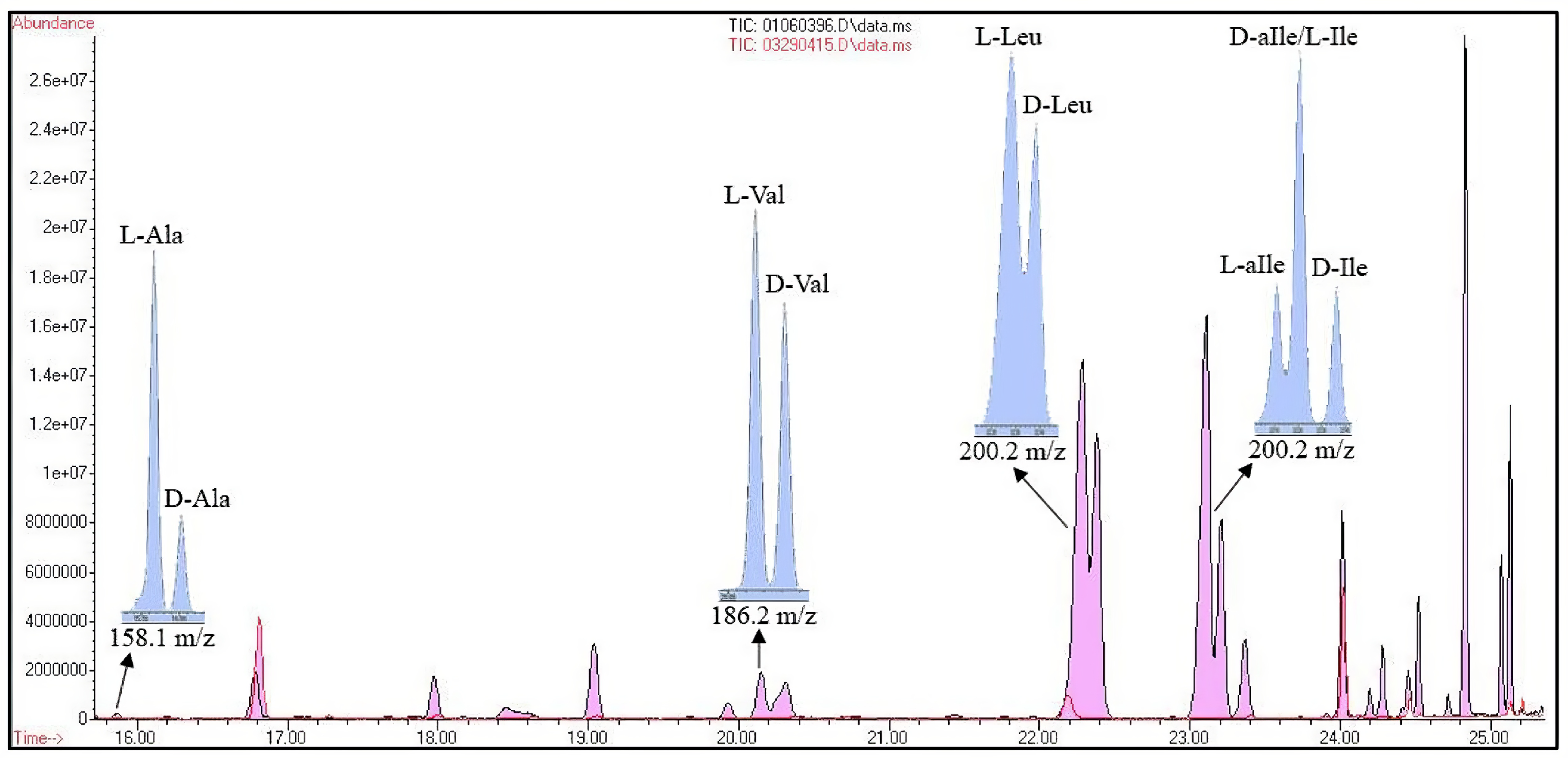
3.1. Determining the Enantiomeric Composition of N-AAA Conjugates Observed in the Case Study
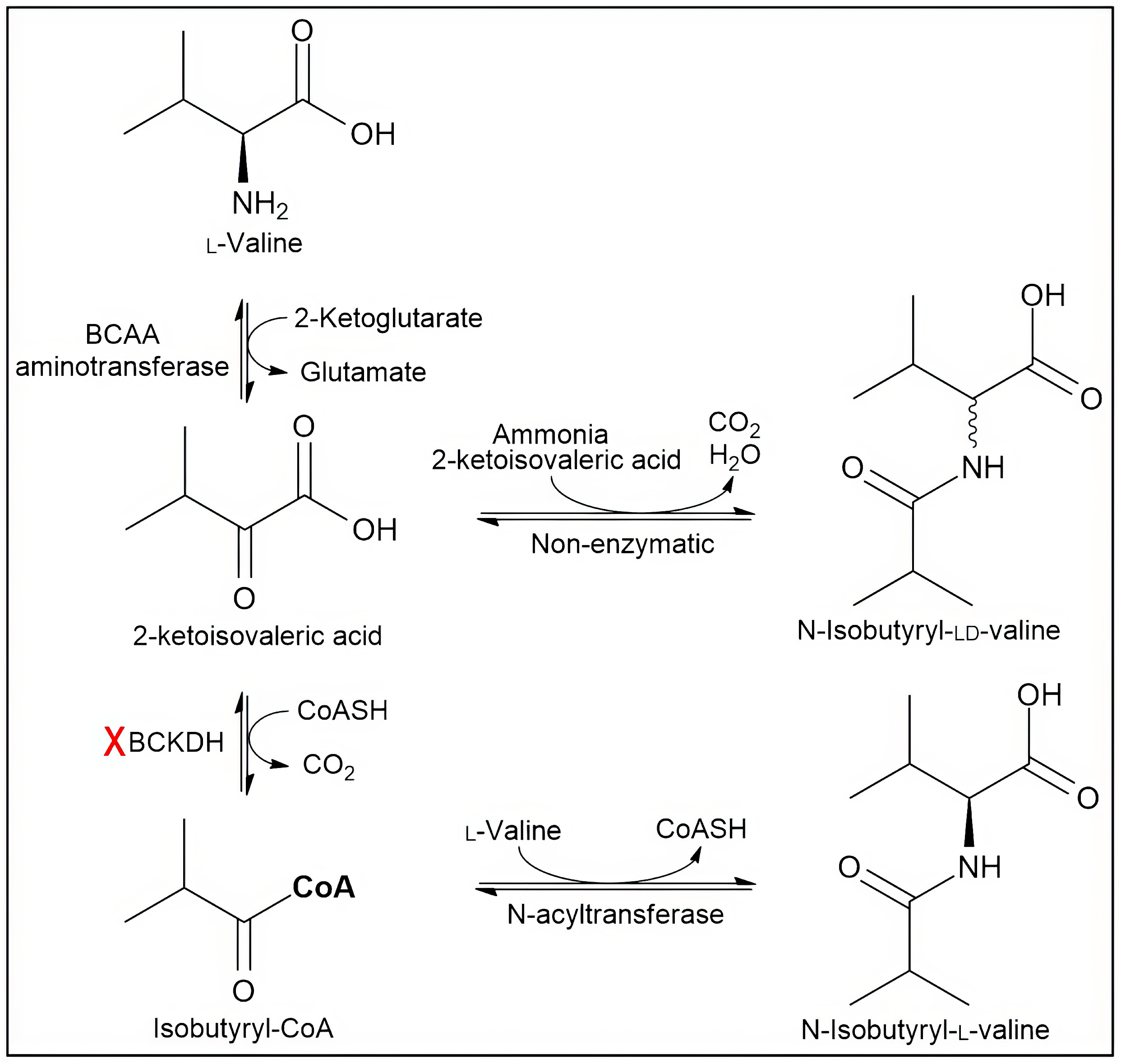
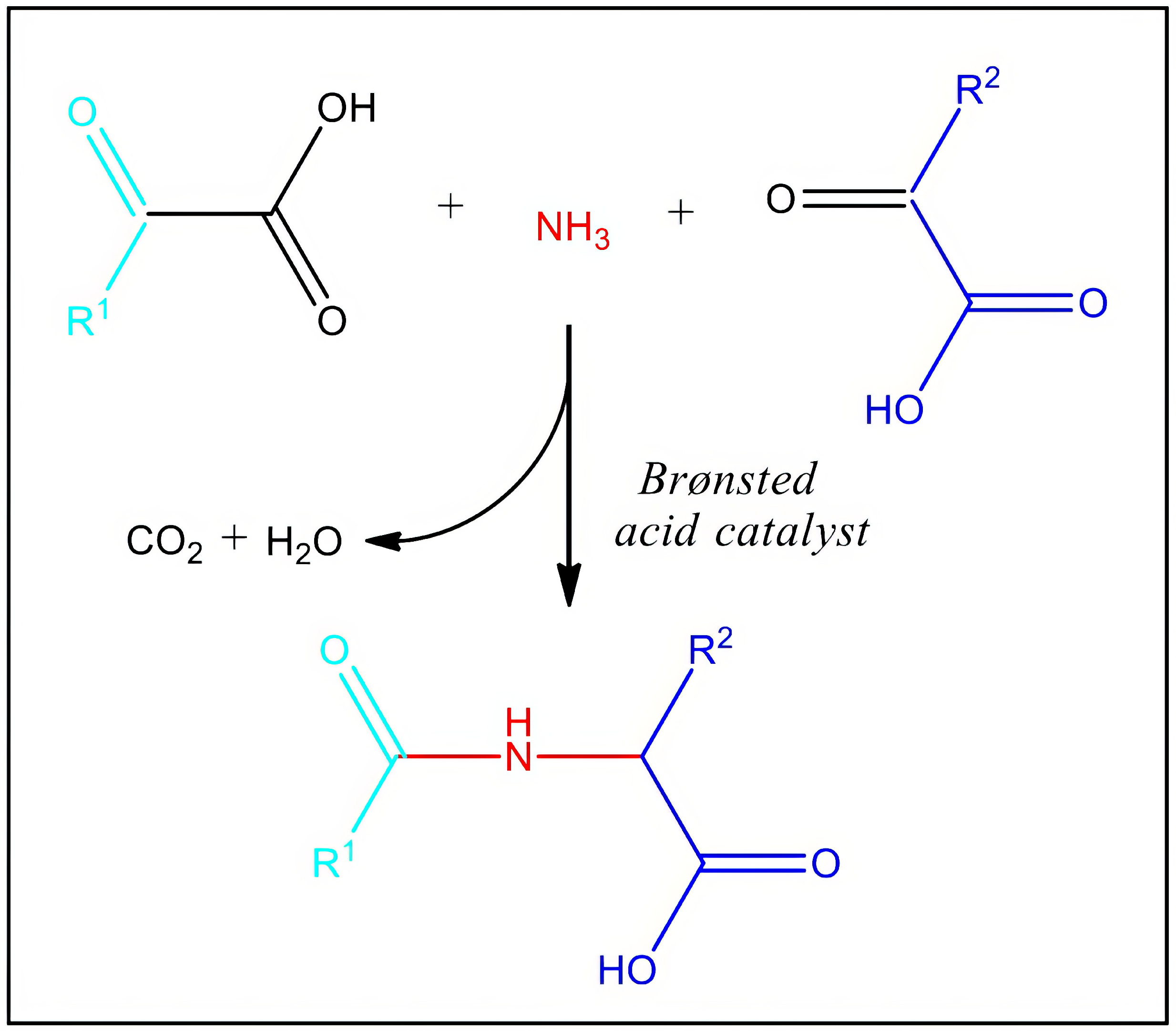
3.2. In Vitro Synthesis at Near-Physiological Conditions
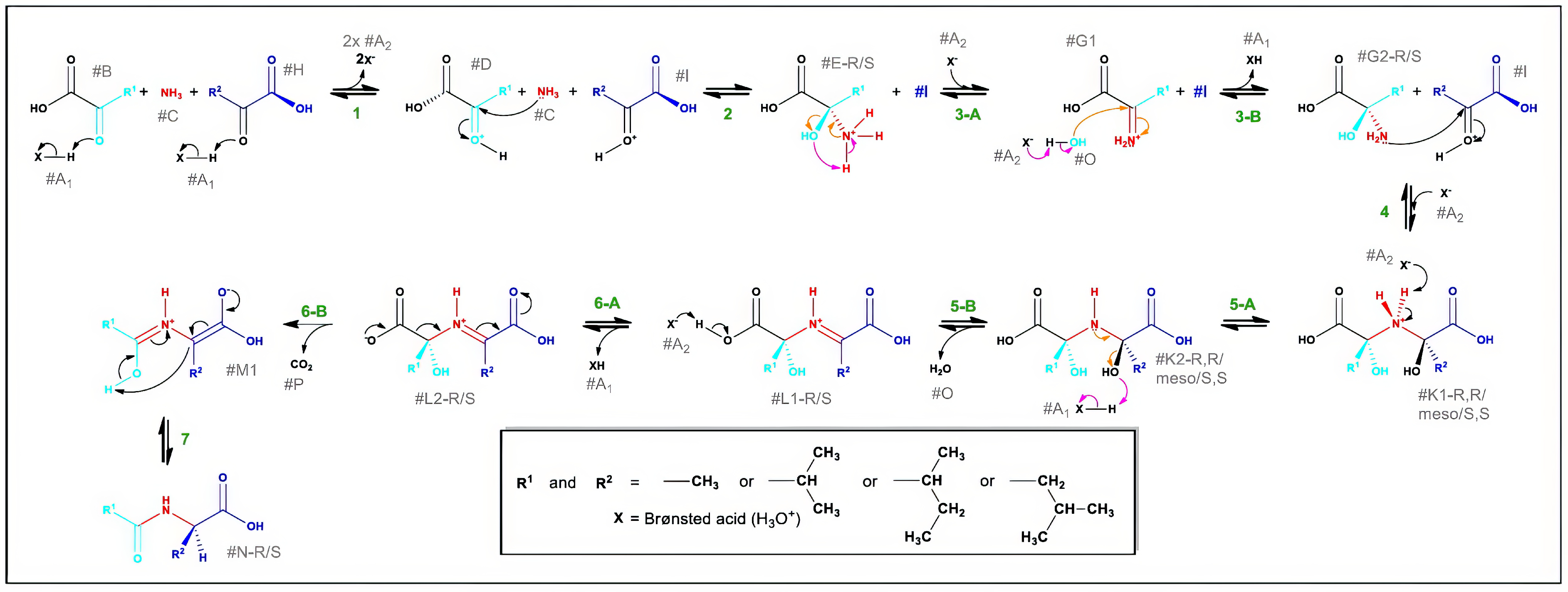
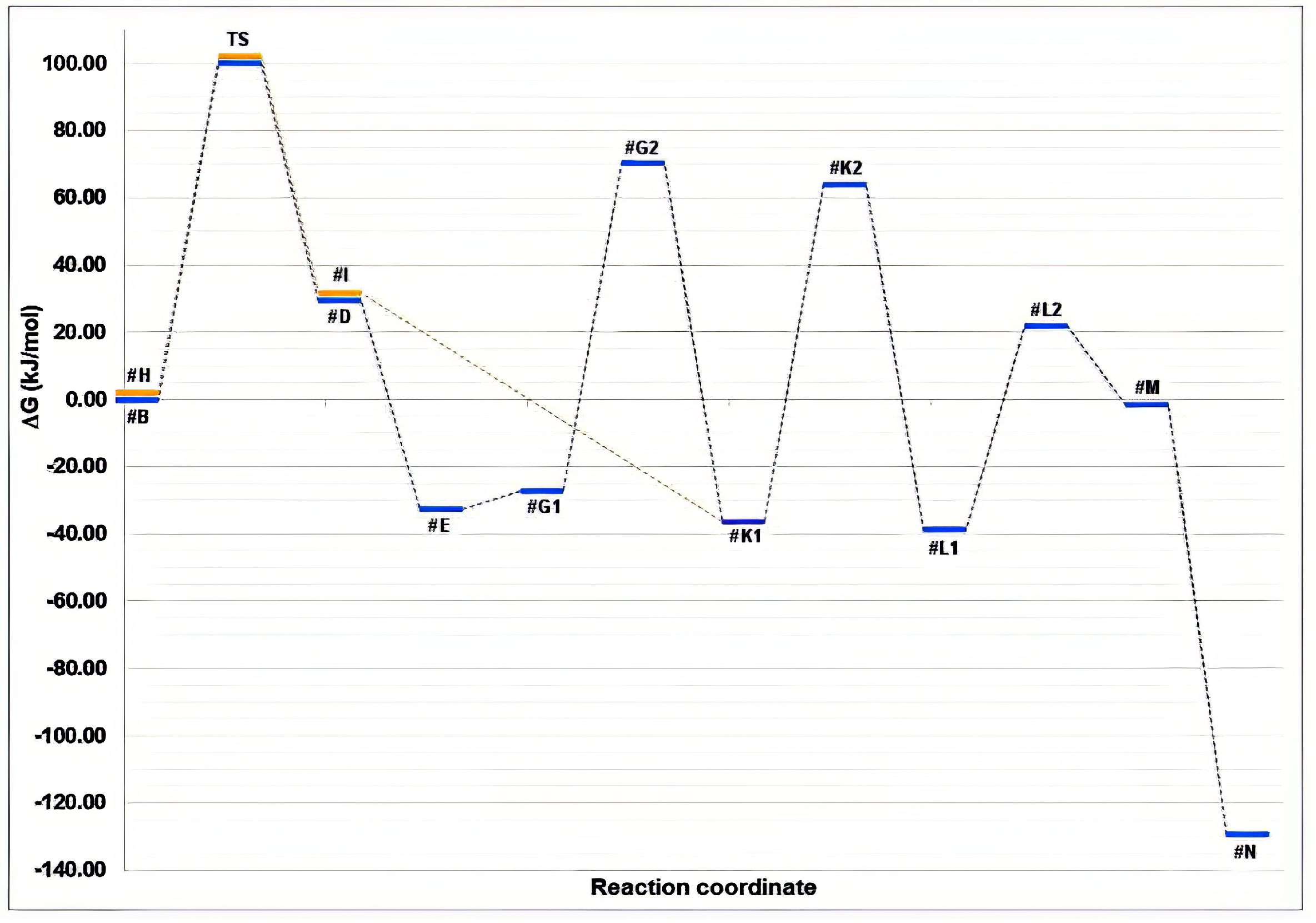
3.3. Molecular Modelling to Investigate the Non-Enzymatic Formation of N-AAA Conjugates
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferreira, C.R.; Van Karnebeek, C.D.M. Inborn errors of metabolism. Handb. Clin. Neurol. 2019, 162, 449–481. [Google Scholar]
- Mordaunt, D.; Cox, D.; Fuller, M. Metabolomics to improve the diagnostic efficiency of inborn errors of metabolism. Int. J. Mol. Sci. 2020, 21, 1195. [Google Scholar]
- Coene, K.L.M.; Kluijtmans, L.A.J.; Van der Heeft, E.; Engelke, U.F.H.; De Boer, S.; Hoegen, B.; Kwast, H.J.T.; Van de Vorst, M.; Huigen, M.C.D.G.; Keularts, I.M.L.W.; et al. Next-generation metabolic screening: Targeted and untargeted metabolomics for the diagnosis of inborn errors of metabolism in individual patients. J. Inherit. Metab. Dis. 2018, 41, 337–353. [Google Scholar]
- Krumsiek, J.; Suhre, K.; Evans, A.M.; Mitchell, M.W.; Mohney, R.P.; Milburn, M.V.; Wägele, B.; Römisch-Margl, W.; Illig, T.; Adamski, J.; et al. mining the unknown: A systems approach to metabolite identification combining genetic and metabolic information. PLoS Genet. 2012, 8, e1003005. [Google Scholar] [CrossRef]
- Peisl, B.Y.L.; Schymanski, E.L.; Wilmes, P. Dark matter in host-microbiome metabolomics: Tackling the unknowns—A review. Anal. Chim. Acta 2018, 1037, 13–27. [Google Scholar]
- Deysel, M.S.M. Biochemiese Karakterisering van ʼn Suid-Afrikaanse MSUD Variant. Master’s Thesis, Potchefstroomse Universiteit vir Christelike Hoër Onderwys, Potchefstroom, South Africa, 2001. [Google Scholar]
- Dry, J. ʼn Studie van Geïnduseerde Metaboliese Weë Weens ʼn Aangebore Defek van Die Vertakte Ketting-α-Ketosuur Dehidrogenase-Kompleks. Bachelor’s (Honours) Thesis, Potchefstroomse Universiteit vir Christelike Hoër Onderwys, Pothchefstroom, South Africa, 1997. [Google Scholar]
- Menkes, J.H.; Hurst, P.L.; Craig, J.M. A new syndrome: Progressive familial infantile cerebral dysfunction associated with an unusual urinary substance. Pediatrics 1954, 14, 462–467. [Google Scholar] [CrossRef]
- Snyderman, S.E.; Norton, P.M.; Roitman, E.; Holt, L.E., Jr. Maple syrup urine disease, with particular reference to dietotherapy. Pediatrics 1964, 34, 454–472. [Google Scholar] [CrossRef]
- Dancis, J.; Levitz, M.; Miller, S.; Westall, R.G. Maple syrup urine disease. Br. Med. J. 1959, 1, 91–93. [Google Scholar] [CrossRef]
- Meister, A.; Abendschein, P.A. Chromatography of α-Keto Acid 2,4-Dinitrophenylhydrazones and Their Hydrogenation Products. Anal. Chem. 1956, 28, 171–173. [Google Scholar] [CrossRef]
- Guder, C.M. Die Gebruik van Filtreerpapier vir Die Versending van Urine vir Die Opsporing van Aangebore Defekte van Die Metabolisme van Organiese Sure. Master’s Thesis, Potchefstroomse Universiteit vir Christelike Hoër Onderwys, Potchefstroom, South Africa, 1988. [Google Scholar]
- Hagenfeldt, L.; Naglo, A.S. New conjugated urinary metabolites in intermediate type maple syrup urine disease. Clin. Chim. Acta 1987, 169, 77–83. [Google Scholar] [CrossRef]
- Schäfer, G.; Bode, J.W. The synthesis of sterically hindered amides. Chimia 2014, 68, 252–255. [Google Scholar] [CrossRef] [PubMed]
- Caldovic, L.; Tuchman, M. N-acetylglutamate and its changing role through evolution. Biochem. J. 2003, 372, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Perrier, J.; Durand, A.; Giardina Puigserver, T.A. Catabolism of intracellular N-terminal acetylated proteins: Involvement of acylpeptide hydrolase and acylase. Biochimie 2005, 87, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Wijayasinghe, Y.S.; Pavlovskym, A.G.; Violam, R.E. Aspartoacylase catalytic deficiency as the cause of Canavan disease: A structural perspective. Biochemistry 2014, 53, 4970–4978. [Google Scholar] [CrossRef] [PubMed]
- Liska, D.; Lyon, M.; Jones, D.S. Detoxification and biotransformational imbalances. Explore 2006, 2, 122–140. [Google Scholar] [PubMed]
- Gerlo, E.; Van Coster, R.; Lissens, W.; Winckelmans, G.; De Meirleir, L.; Wevers, R. Gas chromatographic-mass spectrometric analysis of N-acetylated amino acids: The first case of aminoacylase I deficiency. Anal. Chim. Acta 2006, 571, 191–199. [Google Scholar] [CrossRef]
- Jellum, E.; Horn, L.; Thoresen, O.; Kvittingen, E.A.; Stokke, O. Urinary excretion of N-acetyl amino acids in patients with some inborn errors of amino acid metabolism. Scand. J. Clin. Lab. Suppl. 1986, 184, 21–26. [Google Scholar]
- Sass, J.O.; Mohr, V.; Olbrich, H.; Engelke, U.; Horvath, J.; Fliegauf, M.; Loges, N.T.; Schweitzer-Krantz, S.; Moebus, R.; Weiler, P.; et al. Mutations in ACY1, the gene encoding aminoacylase 1, cause a novel inborn error of metabolism. Am. J. Hum. Genet. 2006, 78, 401–409. [Google Scholar]
- Van der Westhuizen, F.H.; Pretorius, P.J.; Erasmus, E. The utilization of alanine, glutamic acid, and serine as amino acid substrates for glycine N-acyltransferase. J. Biochem. Mol. Toxicol. 2000, 14, 102–109. [Google Scholar]
- Tanaka, K.; Isselbacher, K.J. The Isolation and Identification of N-Isovalerylglycine from Urine of Patients with Isovaleric Acidemia. J. Biol. Chem. 1967, 242, 2966–2972. [Google Scholar]
- Rasmussen, K.; Ando, T.; Nyhan, W.L.; Hull, D.; Cottom, D.; Donnell, G.; Wadlington, W.; Kilroy, A.W. Excretion of Propionylglycine in Propionic Acidaemia. Clin. Sci. 1972, 42, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Sweetman, L.; Weyler, W.; Nyhan, W.L.; De Céspedes, C.; Loria, A.R.; Estrada, Y. Abnormal metabolites of isoleucine in a patient with propionyl-CoA carboxylase deficiency. Biomed. Mass. Spectrom. 1978, 5, 198–207. [Google Scholar] [PubMed]
- Gompertz, D.; Saudubray, J.M.; Charpentier, C.; Bartlett, K.; Goodey, P.A.; Draffan, G.H. A defect in l-isoleucine metabolism associated with alpha-methyl-beta-hydroxybutyric and alpha-methylacetoacetic aciduria: Quantitative in vivo and in vitro studies. Clin. Chim. Acta 1974, 57, 269–281. [Google Scholar] [CrossRef]
- Sweetman, L.; Bates, S.P.; Hull, D.; Nyhan, W.L. Propionyl-CoA carboxylase deficiency in a patient with biotin-responsive 3-methylcrotonylglycinuria. Pediatr. Res. 1977, 11, 1144–1147. [Google Scholar] [CrossRef] [PubMed]
- Rinaldo, P.; O’Shea, J.J.; Coates, P.M.; Hale, D.E.; Stanley, C.A.; Tanaka, K. Medium-chain acyl-CoA dehydrogenase deficiency. Diagnosis by stable-isotope dilution measurement of urinary n-hexanoylglycine and 3-phenylpropionylglycine. N. Engl. J. Med. 1988, 319, 1308–1313. [Google Scholar]
- Goodman, S.I.; McCabe, E.R.B.; Fennessey, P.V.; Mace, J.W. Multiple acyl-coa dehydrogenase deficiency (glutaric aciduria type ii) with transient hypersarcosinemia and sarcosinuria; possible inherited deficiency of an electron transfer flavoprotein. Pediatr. Res. 1980, 14, 12–17. [Google Scholar] [CrossRef]
- Loots, D.T.; Erasmus, E.; Mienie, J. Identification of 19 New Metabolites Induced by Abnormal Amino Acid Conjugation in Isovaleric Acidemia. Clin. Chem. 2005, 51, 1510–1512. [Google Scholar]
- Yanagawa, H.; Makino, Y.; Sato, K.; Nishizawa, M.; Egami, F. Novel formation of alpha-amino acids and their derivatives from oxo acids and ammonia in an aqueous medium. J. Biochem. 1982, 91, 2087–2090. [Google Scholar] [CrossRef]
- Blau, N.; Duran, M.; Gibson, K.M. Laboratory Guide to the Methods in Biochemical Genetics, 1st ed.; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Aksoy, Y.; Ogüs, I.H.; Oauzer, N. Purification and some properties of human placental glucose-6-phosphate dehydrogenase. Protein Expr. Purif. 2001, 21, 286–292. [Google Scholar]
- Cherepanov, A.V.; De Vries, S. Kinetics and thermodynamics of nick sealing by T4 DNA ligase. Eur. J. Biochem. 2003, 270, 4315–4325. [Google Scholar]
- Craig, D.B.; Arriaga, E.A.; Wong, J.C.Y.; Lu, H.; Dovichi, N.J. Studies on Single Alkaline Phosphatase Molecules: Reaction Rate and Activation Energy of a Reaction Catalyzed by a Single Molecule and the Effect of Thermal Denaturation—The Death of an Enzyme. J. Am. Chem. Soc. 1996, 118, 5245–5253. [Google Scholar] [CrossRef]
- Fidaleo, M.; Lavecchia, R. Kinetic Study of Enzymatic Urea Hydrolysis in the pH Range 4–9. Chem. Biochem. Eng. Q. 2003, 17, 311–318. [Google Scholar]
- Kelmer-Bracht, A.M.; Santos, C.P.; Ishii-Iwamoto, E.L.; Broetto-Biazon, A.C.; Bracht, A. Kinetic properties of the glucose 6-phosphatase of the liver from arthritic rats. Biochim. Biophys. Acta 2003, 1638, 50–56. [Google Scholar]
- Matschinsky, F.M.; Zelent, B.; Doliba, N.; Li, C.; Vanderkooi, J.M.; Naji, A.; Sarabu, R.; Grimsby, J. Glucokinase activators for diabetes therapy: May 2010 status report. Diabetes Care 2011, 34, S236–S243. [Google Scholar] [PubMed]
- Scrutton, M.C. Pyruvate Carboxylase: Studies of activator-independent catalysis and of the specificity of activation by acyl derivatives of coenzyme a for the enzyme from rat liver. J. Biol. Chem. 1974, 249, 7057–7067. [Google Scholar] [CrossRef]
- Su, J.-T.; Kim, S.-H.; Yan, Y.-B. Dissecting the pretransitional conformational changes in aminoacylase I thermal denaturation. Biophys. J. 2007, 92, 578–587. [Google Scholar] [PubMed]
- Van Zyl, L.J.; Schubert, W.-D.; Tuffin, M.I.; Cowan, D.A. Structure and functional characterization of pyruvate decarboxylase from Gluconacetobacter diazotrophicus. BMC Struct. Biol. 2014, 14, 21. [Google Scholar]
- Zorich, N.; Jonas, A.; Pownall, H.J. Activation of lecithin cholesterol acyltransferase by human apolipoprotein E in discoidal complexes with lipids. J. Biol. Chem. 1985, 260, 8831–8837. [Google Scholar]
- Keller, M.A.; Piedrafita, G.; Ralser, M. The widespread role of non-enzymatic reactions in cellular metabolism. Curr. Opin. Biotechnol. 2015, 34, 153–161. [Google Scholar]
- Mazaleuskaya, L.L.; Sangkuhl, K.; Thorn, C.F.; FitzGerald, G.A.; Altman, R.B.; Klein, T.E. PharmGKB summary: Pathways of acetaminophen metabolism at the therapeutic versus toxic doses. Pharmacogenet. Genom. 2015, 25, 416–426. [Google Scholar]
- Cava, F.; Lam, H.; De Pedro, M.A.; Waldor, M.K. Emerging knowledge of regulatory roles of D-amino acids in bacteria. Cell. Mol. Life Sci. 2011, 68, 817–831. [Google Scholar] [PubMed]
- Mc Murry, J. Organic Chemistry, 6th ed.; Thomsom Brooks/Cole: Belmount, MA, USA, 2004. [Google Scholar]
- Department of Health Republic of South Africa. Ethics in Health Research: Principle, Processes and Structures, 2nd ed.; Department of Health Republic of South Africa: Cape Town, South Africa, 2015.
- Jacobs, J. Non-Enzymatic Formation of N-Acylated Amino Acid Conjugates in Urine. Master’s Thesis, North-West University, Pothchefstroom, South Africa, 2018. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2-Keto Acid | Expected Result | Actual Product | Yield (μmol/L) |
|---|---|---|---|
| 2-Keto-3-methylvaleric acid | N-2-Methylbutyryl-Ile | N-2-Methylbutyry-Ile | Trace~0.6 |
| 2-Ketobutyric acid | N-Propionyl-2-aminobutyric acid | N-Propionyl-2-aminobutyric acid | 10.12 |
| 2-Ketocaproic acid | N-Pentanoyl-nor-Leu | N-Pentanoyl-norleucine | 14.70 |
| 2-Ketoisocaproic acid | N-Isovaleryl-Leu | N-Isovaleryl-Leu | 1.35 |
| 2-Ketoisovaleric acid | N-Isobutyryl-Val | N-Isobutyryl-Val | 20.32 |
| 2-Ketovaleric acid | N-Butyryl-Nva | N-Butyryl-Nva | Trace~1.0 |
| Phenylpyruvate | N-(Phenylacetyl)-Phe | N-(Phenylacetyl)-Phe | 5.22 |
| p-Hydroxyphenylpyruvate | N-(p-hydroxyphenylacetyl)-(p-hydroxy-Phe) | No result | Not detected |
| Pyruvate | N-Ac-Ala | N-Ac-Ala | 2.46 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacobs, J.; van Sittert, C.G.C.E.; Mienie, L.J.; Dercksen, M.; Opperman, M.; Vorster, B.C. Non-Enzymatic Formation of N-acetylated Amino Acid Conjugates in Urine. Appl. Sci. 2023, 13, 10002. https://doi.org/10.3390/app131810002
Jacobs J, van Sittert CGCE, Mienie LJ, Dercksen M, Opperman M, Vorster BC. Non-Enzymatic Formation of N-acetylated Amino Acid Conjugates in Urine. Applied Sciences. 2023; 13(18):10002. https://doi.org/10.3390/app131810002
Chicago/Turabian StyleJacobs, Jano, Cornelia Gertina Catharina Elizabeth van Sittert, Lodewyk Japie Mienie, Marli Dercksen, Monique Opperman, and Barend Christiaan Vorster. 2023. "Non-Enzymatic Formation of N-acetylated Amino Acid Conjugates in Urine" Applied Sciences 13, no. 18: 10002. https://doi.org/10.3390/app131810002
APA StyleJacobs, J., van Sittert, C. G. C. E., Mienie, L. J., Dercksen, M., Opperman, M., & Vorster, B. C. (2023). Non-Enzymatic Formation of N-acetylated Amino Acid Conjugates in Urine. Applied Sciences, 13(18), 10002. https://doi.org/10.3390/app131810002