
Modification of 6,7-Dichloro-5,8-Quinolinedione at C2 Position: Synthesis, Quantum Chemical Properties, and Activity against DT-Diaphorase Enzyme

Abstract

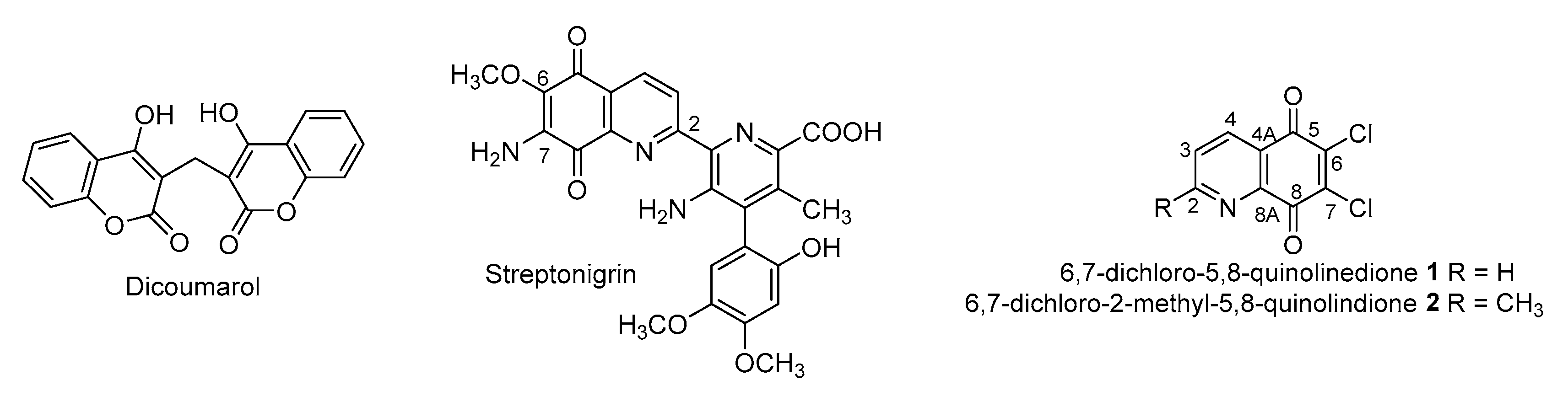
1. Introduction
2. Materials and Methods
2.1. Physical Measurements
2.2. Chemistry
2.2.1. Synthesis of 6,7-Dichloro-5,8-Dioxo-5,8-Dihydroquinoline-2-Carbaldehyde 3
2.2.2. Synthesis of 2-Substituted-5,8-Quinolinedione 6–7
2.3. Computational Details
2.4. Enzymatic Assay
2.5. Molecular Docking Study
3. Results and Discussion
3.1. Chemistry
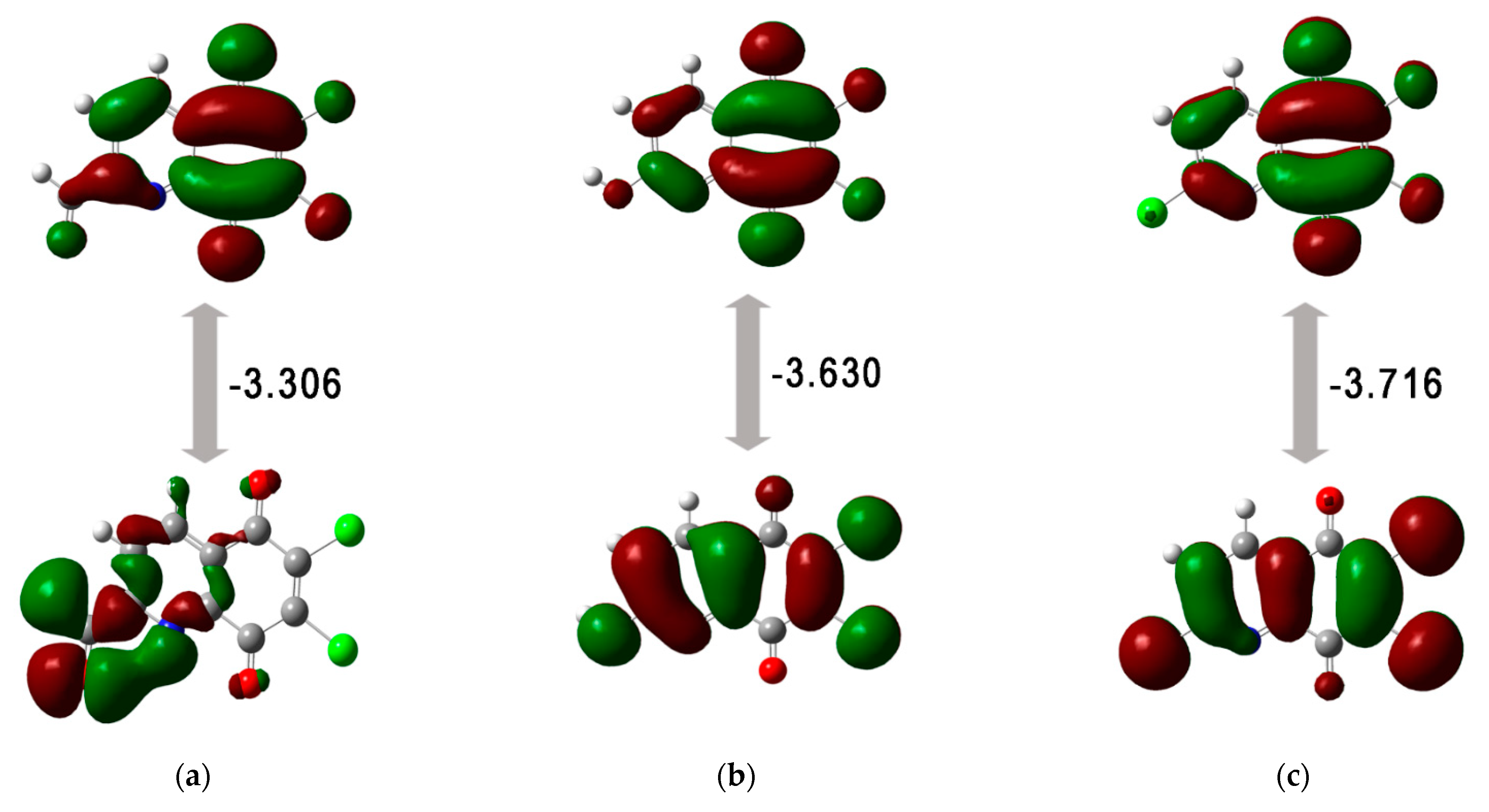
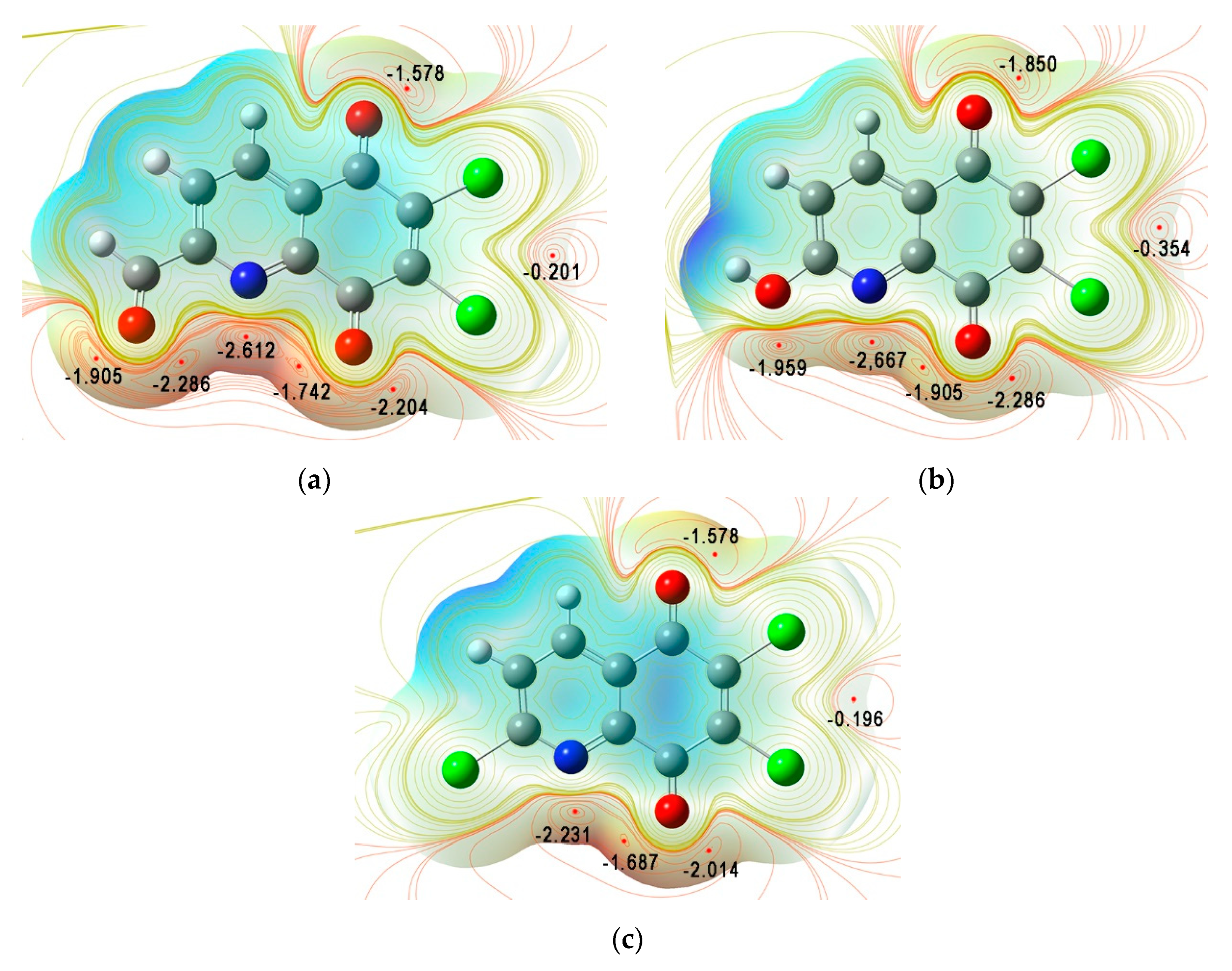
3.2. Quantum Chemical Properties
3.3. ADMET Analysis
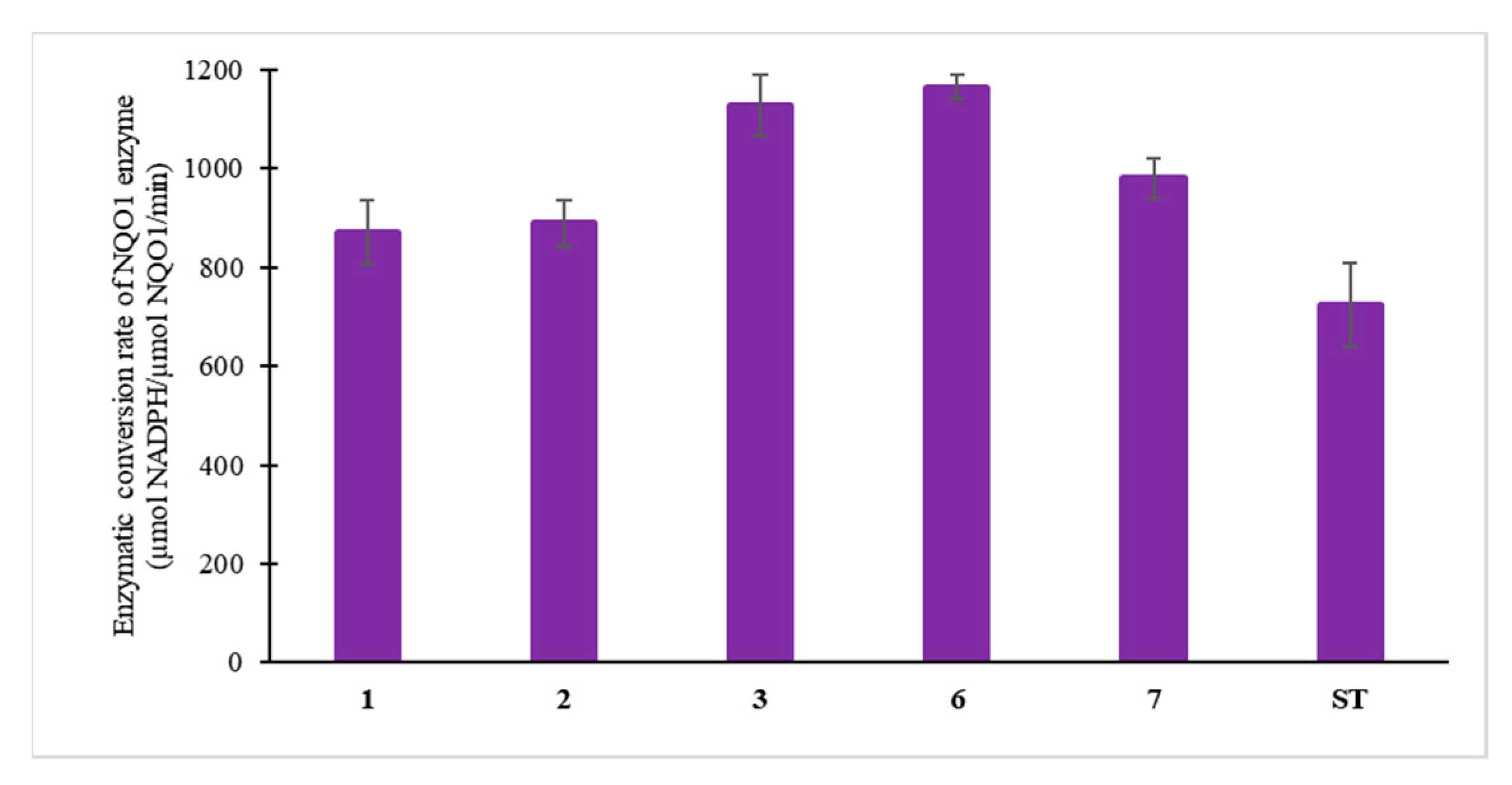
3.4. Enzymatic Study
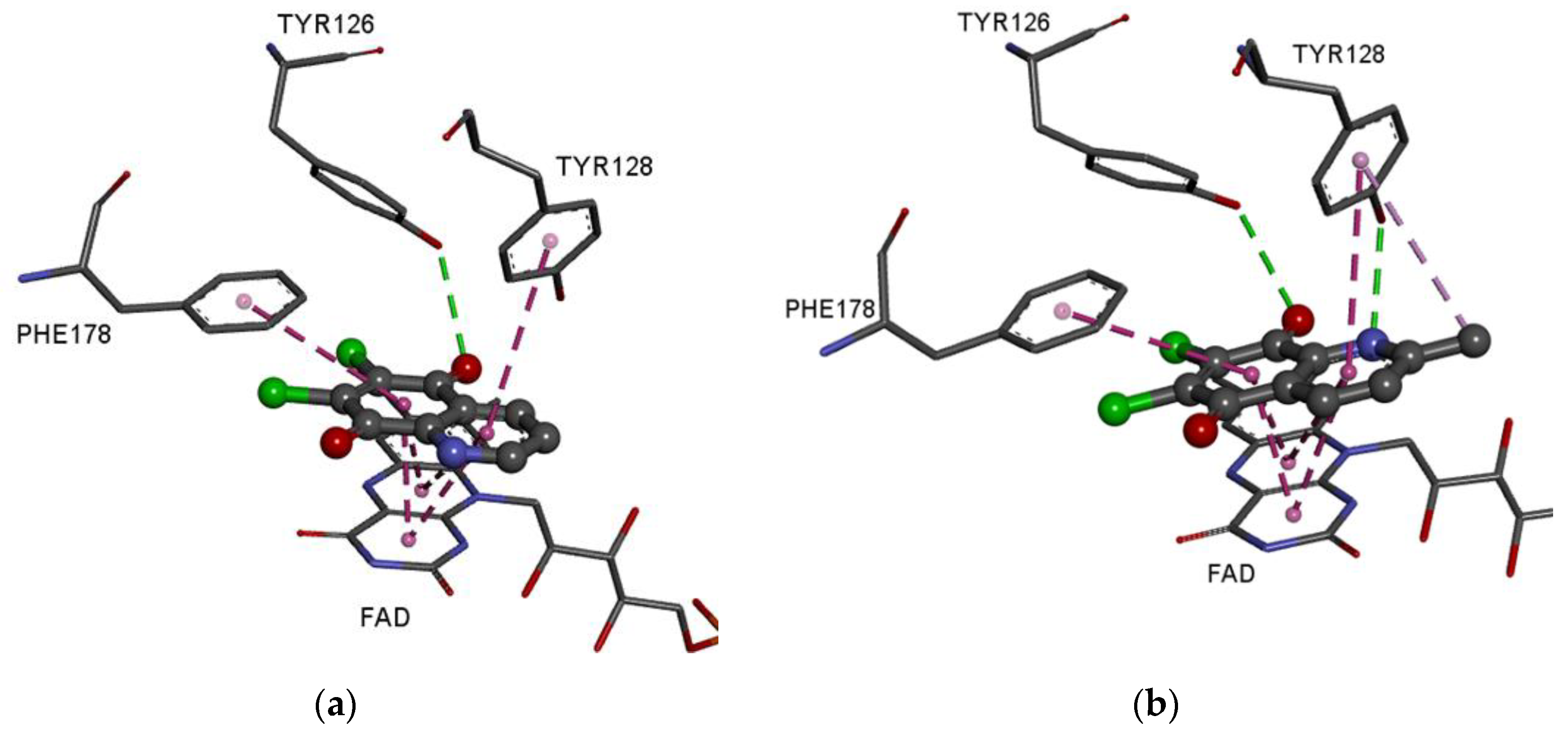
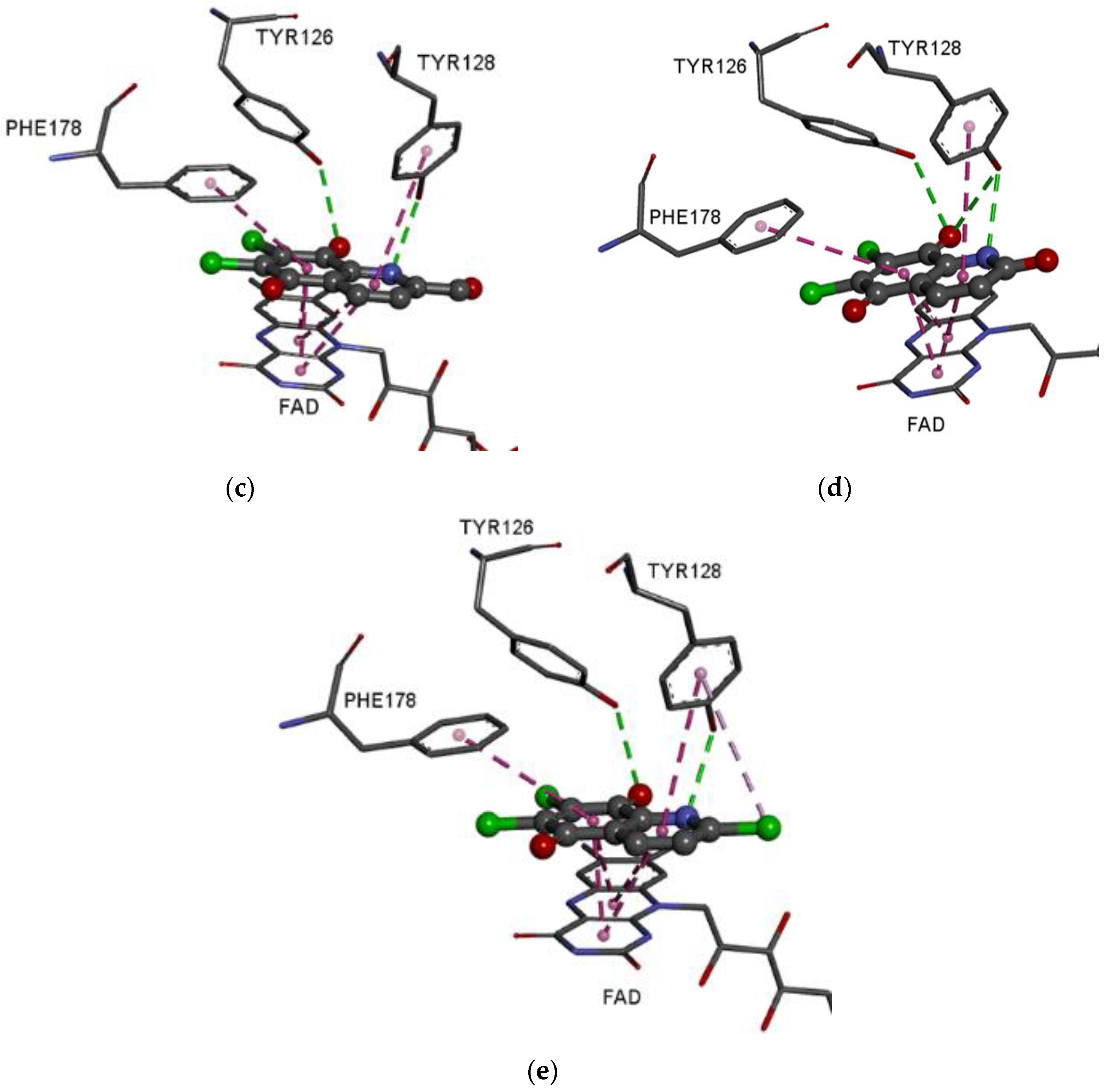
3.5. Molecular Docking Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ernster, L. Diaphorase activities in liver ctoplasmic fractions. Fed. Proc. 1958, 17, 216. [Google Scholar]
- Ross, D.; Siegel, D. The diverse functionality of NQO1 and its roles in redox control. Redox Biol. 2021, 41, 101950. [Google Scholar] [CrossRef]
- Beaver, S.K.; Mesa-Torres, N.; Pey, A.L.; Timson, D.J. NQO1: A target for the treatment of cancer and neurological diseases, and a model to understand loss of function disease mechanisms. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.; Ross, D. Immunodetection of NAD(P)H:quinone oxidoreductase 1 (NQO1) in human tissues. Free Radic. Biol. Med. 2000, 29, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.; Franklin, W.A.; Ross, D. Immunohistochemical detection of NAD(P)H:quinone oxidoreductase in human lung and lung tumors. Clin. Cancer Res. 1998, 4, 2065–2070. [Google Scholar]
- Strassburg, A.; Strassburg, C.P.; Manns, M.P.; Tukey, R.H. Differential gene expression of NAD(P)H:quinone oxidoreductase and NRH:quinone oxidoreductase in human hepatocellular and biliary tissue. Mol. Pharmacol. 2002, 61, 320–325. [Google Scholar] [CrossRef]
- Schor, N.A.; Morris, H. The activity of the DT-diaphorase in experimental hepatomas. Cancer Biochem. Biophys. 1977, 2, 5–9. [Google Scholar]
- Faig, M.; Bianchet, M.; Winski, S.; Hargreaves, R.; Moody, C.; Hudnott, A.; Ross, D.; Amzel, L. Structure-based development of anticancer drugs: Complexes of NAD(P)H:quinone oxidoreductase 1 with chemotherapeutic quinones. Structure 2001, 9, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Bianchet, M.A.; Talalay, P.; Amzel, L.M. The three-dimensional structure of NAD(P)H:quinone reductase, a flavoprotein involved in cancer chemoprotection and chemotherapy: Mechanism of the two-electron reduction. Proc. Natl. Acad. Sci. USA 1995, 92, 8846–8850. [Google Scholar] [CrossRef]
- Iyanagi, T.; Yamazaki, I. One-electron-transfer reactions in biochemical systems. V. Difference in the mechanism of quinone reduction by the NADH dehydrogenase and the NAD(P)H dehydrogenase (DT-diaphorase). Biochim. Biophys. Acta 1970, 216, 282–294. [Google Scholar] [CrossRef]
- Tedeschi, G.; Chen, S.; Massey, V. DT-diaphorase. Redox potential, steady-state, and rapid reaction studies. J. Biol. Chem. 1995, 270, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Belinsky, M.; Jaiswal, A.K. NAD(P)H:quinone oxidoreductase1 (DT-diaphorase) expression in normal and tumor tissues. Cancer Metastasis Rev. 1993, 12, 103–117. [Google Scholar] [CrossRef]
- Wu, L.Q.; Ma, X.; Zhang, C.; Liu, Z.P. Design, synthesis, and biological evaluation of 4-substituted-3,4-dihydrobenzo[h]quinoline-2,5,6(1H)-triones as NQO1-directed antitumor agents. Eur. J. Med. Chem. 2020, 198, 112396. [Google Scholar] [CrossRef] [PubMed]
- Timson, D.J. Dicoumarol: A drug which hits at least two very different targets in vitamin K metabolism. Curr. Drug Targets 2017, 18, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Cullen, J.J.; Hinkhouse, M.M.; Grady, M.; Gaut, A.W.; Liu, J.; Zhang, Y.P.; Weydert, C.J.; Domann, F.E.; Oberley, L.W. Dicumarol inhibition of NADPH:quinone oxidoreductase induces growth inhibition of pancreatic cancer via a superoxide-mediated mechanism. Cancer Res. 2003, 63, 5513–5520. [Google Scholar] [PubMed]
- Lewis, A.; Ough, M.; Li, L.; Hinkhouse, M.M.; Ritchie, J.M.; Spitz, D.R.; Cullen, J.J. Treatment of pancreatic cancer cells with dicumarol induces cytotoxicity and oxidative stress. Clin. Cancer Res. 2004, 10, 4550–4558. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.; Yan, C.; Ross, D. NAD(P)H:quinone oxidoreductase 1 (NQO1) in the sensitivity and resistance to antitumor quinones. Biochem. Pharmacol. 2012, 83, 1033–1040. [Google Scholar] [CrossRef]
- Bolzán, A.D.; Bianchi, M.S. Genotoxicity of streptonigrin: A review. Mutat. Res. 2001, 488, 25–37. [Google Scholar] [CrossRef]
- Bringmann, G.; Reichert, Y.; Kane, V. The total synthesis of streptonigrin and related antitumor antibiotic natural products. Tetrahedron 2004, 30, 3539–3574. [Google Scholar] [CrossRef]
- Donohoe, T.J.; Jones, C.R.; Kornahrens, A.F.; Barbosa, L.C.A.; Walport, L.J.; Tatton, M.R.; O’Hagan, M.; Rathi, A.H.; Baker, D.B. Total synthesis of the antitumor antibiotic (±)-streptonigrin: First- and second-generation routes for de novo pyridine formation using ring-closing metathesis. J. Org. Chem. 2013, 78, 12338–12350. [Google Scholar] [CrossRef] [PubMed]
- Kadela-Tomanek, M.; Bębenek, E.; Chrobak, E.; Boryczka, S. 5,8-Quinolinedione scaffold as a promising moiety of bioactive agents. Molecules 2019, 24, 4115. [Google Scholar] [CrossRef] [PubMed]
- Kadela-Tomanek, M.; Jastrzębska, M.; Chrobak, E.; Bębenek, E.; Latocha, M. Hybrids of 1,4-quinone with quinoline derivatives: Synthesis, biological activity, and molecular docking with DT-Diaphorase (NQO1). Molecules 2022, 27, 6206. [Google Scholar] [CrossRef] [PubMed]
- Kadela-Tomanek, M.; Jastrzębska, M.; Marciniec, K.; Chrobak, E.; Bębenek, E.; Latocha, M.; Kuśmierz, D.; Boryczka, S. Design, synthesis and biological activity of 1,4-quinone moiety attached to betulin derivatives as potent DT-diaphorase substrate. Bioorg. Chem. 2021, 106, 104478. [Google Scholar] [CrossRef]
- Mulchin, B.; Newton, C.G.; Baty, J.W.; Grasso, C.H.; Martin, W.J.; Walton, M.C.; Dangerfield, E.M.; Plunkett, C.H.; Berridge, M.V.; Harper, J.L.; et al. The anti-cancer, anti-inflammatory and tuberculostatic activities of a series of 6,7-substituted-5,8-quinolinequinones. Bioorg. Med. Chem. 2010, 19, 3238–3251. [Google Scholar] [CrossRef]
- Ling, Y.; Yang, Q.; Teng, Y.; Chen, S.; Gao, W.; Guo, J.; Hsu, P.; Liu, Y.; Morris-Natschke, S.; Hung, C.; et al. Development of novel amino-quinoline-5,8-dione derivatives as NAD(P)H:quinone oxidoreductase 1 (NQO1) inhibitors with potent antiproliferative activities. Eur. J. Med. Chem. 2018, 154, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Ryu, C.; Oh, S.; Choi, S.; Kang, D. Synthesis of antifungal evaluation of 2H-[1,2,3]Triazolo [4,5-g]isoquinoline-4,9-diones. Chem. Pharm. Bull. 2014, 62, 1119–1124. [Google Scholar] [CrossRef]
- Yoo, K.; Yoon, E.; Park, Y.; Park, S.; Lee, C.; Lee, W.; Chi, D.; Kim, D. Synthesis and SAR of aziridinylquinoline-5,8-diones as agents against malignant tumor cells. Bull. Korean Chem. Soc. 2001, 22, 1067–1068. [Google Scholar]
- Kadela-Tomanek, M.; Pawełczak, B.; Jastrzębska, M.; Bębenek, E.; Chrobak, E.; Latocha, M.; Kusz, J.; Książek, M.; Boryczka, S. Structural, vibrational and quantum chemical investigations for 6,7-dichloro-2-methyl-5,8-quinolinedione. Cytotoxic and molecular docking studies. J. Mol. Struct. 2018, 1168, 73–83. [Google Scholar] [CrossRef]
- Jianbo, H.; Tingting, Z.; Yongjing, C.; Yuanyuan, Z.; Weiqing, Y.; Menglin, M. Study on relationship between fluorescence properties and structure of substituted 8-hydroxyquinoline zinc complexes. J. Fluoresc. 2018, 28, 1121–1126. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16; Revision A. 03. 2016; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Wolinski, K.; Hinton, J.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Foresman, J.; Frisch, E. Exploring Chemistry with Electronic Structure Methods: A Guide to Using Gaussian; Gaussian: Pittsburg, PA, USA, 1996. [Google Scholar]
- Politzer, P.; Laurence, P.; Jayasuriya, K. Molecular electrostatic potentials: An effective tool for the elucidation of biochemical phenomena. Environ. Health Perspect. 1985, 61, 191–202. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.; Millam, J. GaussView, Version 5; Semichem Inc.: Shawnee, KS, USA, 2009. [Google Scholar]
- pkCSM. Available online: http://biosig.unimelb.edu.au/pkcsm/prediction# (accessed on 10 January 2023).
- Pires, D.; Blundell, T.; Ascher, D. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Bian, J.; Wang, N.; Qian, X.; Gu, J.; Mu, T.; Fan, J.; Yang, X.; Li, S.; Yang, T.; et al. Novel naphtho [2,1-d]oxazole-4,5-diones as NQO1 substrates with improved aqueous solubility: Design, synthesis, and in vivo antitumor evaluation. Bioorg. Med. Chem. 2016, 24, 1006–1013. [Google Scholar] [CrossRef] [PubMed]
- Bian, J.; Deng, B.; Xu, L.; Xu, X.; Wang, N.; Hu, T.; Yao, Z.; Du, J.; Yang, L.; Lei, Y.; et al. 2-Substituted 3-methylnaphtho [1,2-b]furan-4,5-diones as novel L-shaped ortho-quinone substrates for NAD(P)H:quinone oxidoreductase (NQO1). Eur. J. Med. Chem. 2014, 82, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Dym, O.; Tsvetkov, P.; Adler, J.; Shaul, Y. The crystal structure of NAD(P)H Quinone Oxidoreductase 1 in complex with its potent inhibitor dicoumarol. Biochemistry 2006, 45, 6372–6378. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Dessault Systemes BIOVIA. Available online: https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/ (accessed on 12 December 2022).
- Hehre, W.; Ditchfield, R.; Pople, J. Self—Consistent molecular orbital methods. XII. Further extensions of Gaussian-type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Cheeseman, J.; Trucks, G.; Keith, T.; Frisch, M. A comparison of models for calculating nuclear magnetic resonance shielding tensors. J. Chem. Phys. 1994, 104, 5497–5509. [Google Scholar] [CrossRef]
- Pérez, P.; Domingo, L.R.; Aurell, M.J.; Contreras, R. Quantitative characterization of the global electrophilicity pattern of some reagents involved in 1,3-dipolar cycloaddition reactions. Tetrahedron 2003, 59, 3117–3125. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef]
- Ali, B.; Khalid, M.; Asim, S.; Khan, M.U.; Iqbal, Z.; Hussain, A.; Hussain, R.; Ahmed, S.; Ali, A.; Hussain, A.; et al. Key electronic, linear and nonlinear optical properties of designed disubstituted quinoline with carbazole compounds. Molecules 2021, 26, 2760. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, Y.; Ren, Q.; Yu, D.; Lu, H. Synthesis, crystal structure and DFT study of a novel compound N-(4-(2,4-dimorpholinopyrido [2,3-d]pyrimidin-6-yl)phenyl)pyrrolidine-1-carboxamide. J. Mol. Struct. 2021, 1235, 130261. [Google Scholar] [CrossRef]
- Marya, Y.S.; Mary, J.; Krátký, M.; Vinsov, J.; Baraldi, C.; Gamberini, M. DFT, molecular docking and SERS (concentration and solvent dependant) investigations of a methylisoxazole derivative with potential antimicrobial activity. J. Mol. Struct. 2021, 1232, 130034. [Google Scholar] [CrossRef]
- Chandrasekaran, K.; Kumar, R. Structural, spectral, thermodynamical, NLO, HOMO, LUMO and NBO analysis of fluconazole. Spectrochim. Acta A 2015, 150, 974–991. [Google Scholar] [CrossRef] [PubMed]
- Jakhar, R.; Dangi, M.; Khichi, A.; Chhillar, A.K. Relevance of molecular docking studies in drug designing. Curr. Bioinf. 2019, 15, 270–278. [Google Scholar] [CrossRef]
- Al-Attraqchi, O.H.A.; Venugopala, K.N. 2D- and 3D-QSAR modeling of imidazole-based glutaminyl cyclase inhibitors. Curr. Comput. Aided Drug Des. 2020, 16, 682–697. [Google Scholar] [CrossRef] [PubMed]
- Delaney, J.S. ESOL: Estimating aqueous solubility directly from molecular structure. J. Chem. Inf. Model. 2004, 44, 1000–1005. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proton | 1H NMR δ (ppm) | HSQC | Carbon | 13C NMR δ (ppm) | HMBC |
|---|---|---|---|---|---|
| CHO | 10.35 | 10.35–191.4 | CHO | 191.4 | 10.35–155.9 (C2) 10.35–125.2 (C3) 10.35–137.5 (C4) |
| H4 | 8.77 | 8.77–137.5 | C4 | 137.5 | 8.77–147.0 (C8A) 8.77–155.9 (C2) 8.77–175.0 (C5) |
| H3 | 8.40 | 8.40–125.6 | C3 | 125.2 | 8.40–130.4 (C4A) 8.40–155.8 (C2) 8.40–191.4 (CHO) |
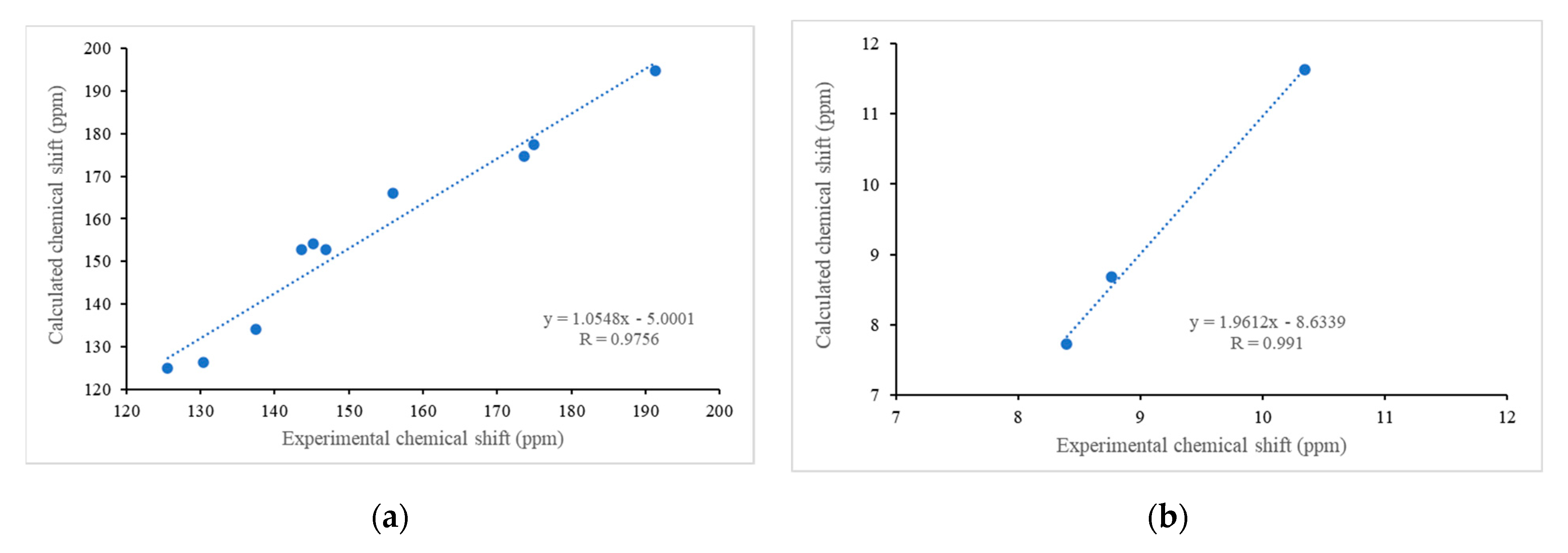
| Atoms | Chemical Shift δ (ppm) | |
|---|---|---|
| Experimental | Calculated | |
| CHO | 10.35 | 11.63 |
| H4 | 8.77 | 8.69 |
| H3 | 8.40 | 7.73 |
| CHO | 191.4 | 194.8 |
| C2 | 155.9 | 166.1 |
| C3 | 125.2 | 124.9 |
| C4 | 137.5 | 134.1 |
| C4A | 130.4 | 126.5 |
| C5 | 175.0 | 177.5 |
| C6 | 143.6 | 152.8 |
| C7 | 145.1 | 154.2 |
| C8 | 173.7 | 174.8 |
| C8A | 147.0 | 152.9 |
| Compound | EHOMO | ELUMO | ΔE | I | A | η | µ | χ | ω |
|---|---|---|---|---|---|---|---|---|---|
| 3 | −7.865 | −4.559 | −3.306 | 7.865 | 4.559 | 1.653 | −6.212 | 6.212 | 11.672 |
| 6 | −7.860 | −4.230 | −3.630 | 7.860 | 4.230 | 1.815 | −6.045 | 6.045 | 10.066 |
| 7 | −8.281 | −4.564 | −3.716 | 8.281 | 4.564 | 1.858 | −6.422 | 6.422 | 11.100 |
| Compound | logS | logP | MW (g/mol) | HA | HD | LC50 (mM) | Hepatotoxicity |
|---|---|---|---|---|---|---|---|
| 1 | −3.08 | 2.14 | 228 | 3 | 0 | 8.710 | No |
| 2 | −3.39 | 2.45 | 242 | 3 | 0 | 6.531 | No |
| 3 | −3.01 | 1.96 | 256 | 4 | 0 | 8.810 | No |
| 6 | −3.04 | 1.86 | 244 | 4 | 1 | 28.249 | No |
| 7 | −3.87 | 2.80 | 262 | 3 | 0 | 8.790 | No |
| Molecules | ΔG (kcal/mol) |
|---|---|
| 1 | −8.1 |
| 2 | −8.0 |
| 3 | −8.3 |
| 6 | −8.3 |
| 7 | −8.2 |
| ST | −7.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kadela-Tomanek, M.; Bębenek, E.; Chrobak, E. Modification of 6,7-Dichloro-5,8-Quinolinedione at C2 Position: Synthesis, Quantum Chemical Properties, and Activity against DT-Diaphorase Enzyme. Appl. Sci. 2023, 13, 1530. https://doi.org/10.3390/app13031530
Kadela-Tomanek M, Bębenek E, Chrobak E. Modification of 6,7-Dichloro-5,8-Quinolinedione at C2 Position: Synthesis, Quantum Chemical Properties, and Activity against DT-Diaphorase Enzyme. Applied Sciences. 2023; 13(3):1530. https://doi.org/10.3390/app13031530
Chicago/Turabian StyleKadela-Tomanek, Monika, Ewa Bębenek, and Elwira Chrobak. 2023. "Modification of 6,7-Dichloro-5,8-Quinolinedione at C2 Position: Synthesis, Quantum Chemical Properties, and Activity against DT-Diaphorase Enzyme" Applied Sciences 13, no. 3: 1530. https://doi.org/10.3390/app13031530
APA StyleKadela-Tomanek, M., Bębenek, E., & Chrobak, E. (2023). Modification of 6,7-Dichloro-5,8-Quinolinedione at C2 Position: Synthesis, Quantum Chemical Properties, and Activity against DT-Diaphorase Enzyme. Applied Sciences, 13(3), 1530. https://doi.org/10.3390/app13031530