
Consequences of Glucose Enriched Diet on Oncologic Patients




Abstract
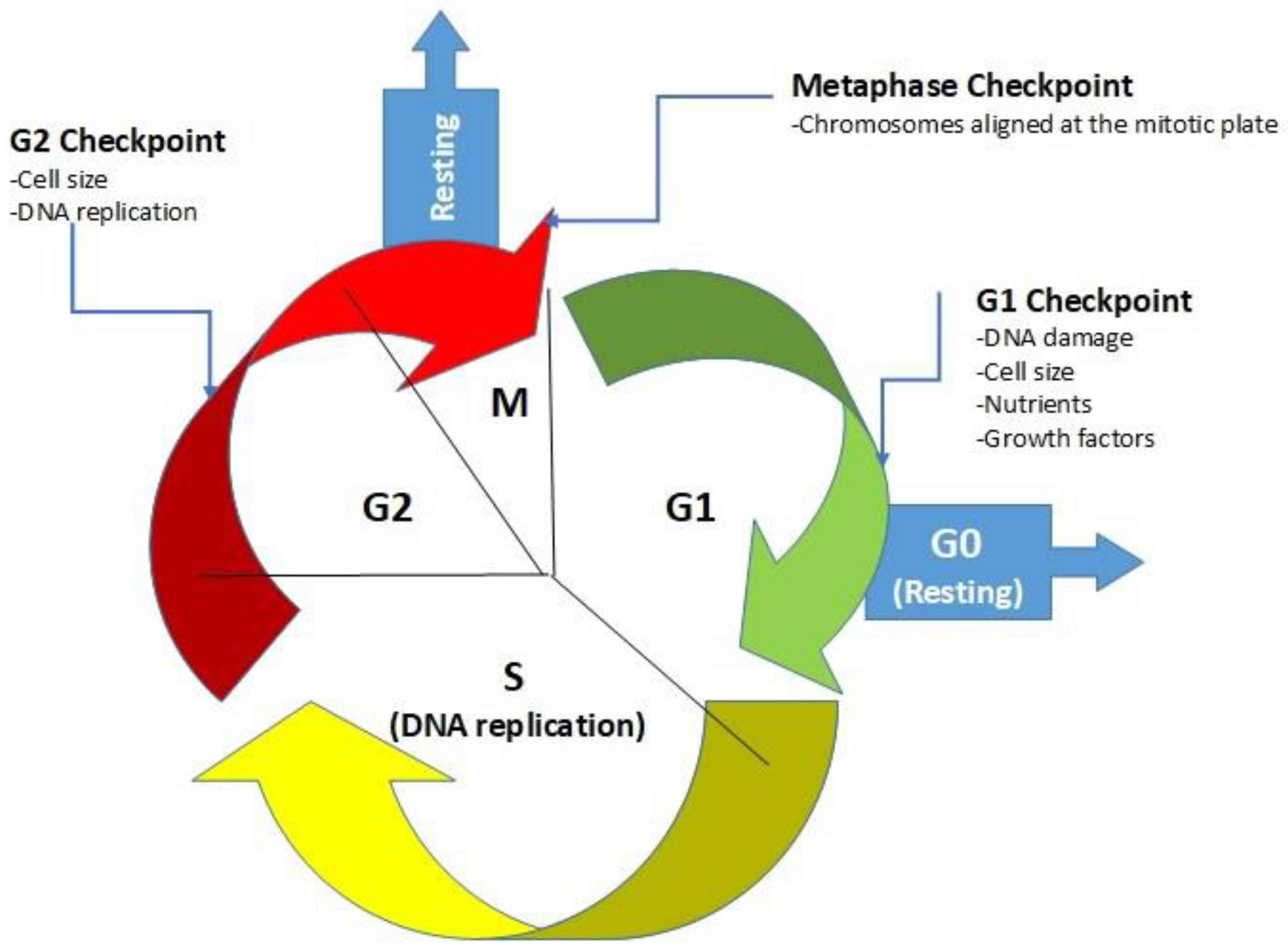
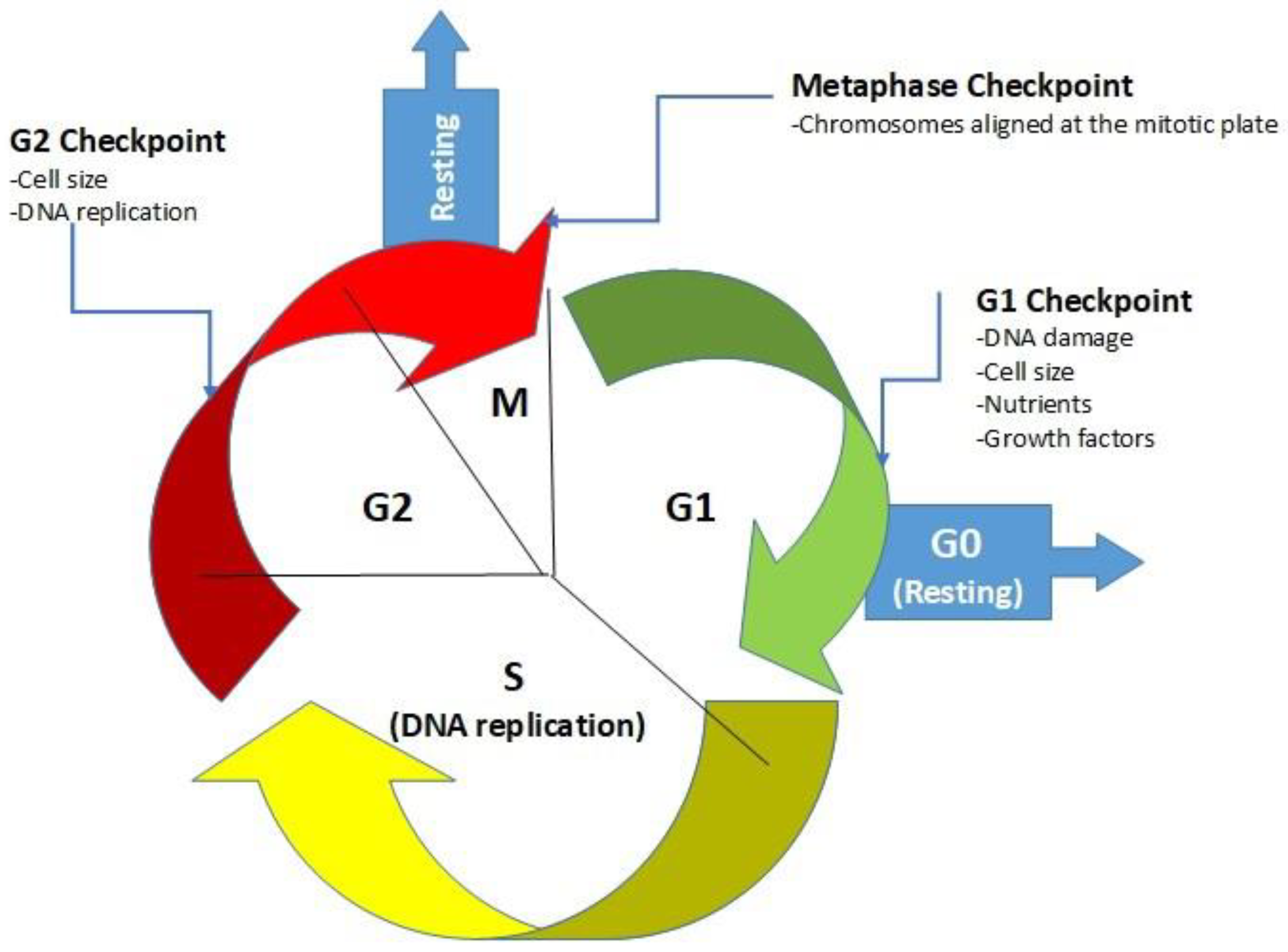
:1. Introduction
1.1. Glucose and Its Influence on Cancer
1.2. Role of Sugar in Cancer Metabolism
1.2.1. Sugars in the Diet and Cancer Risk
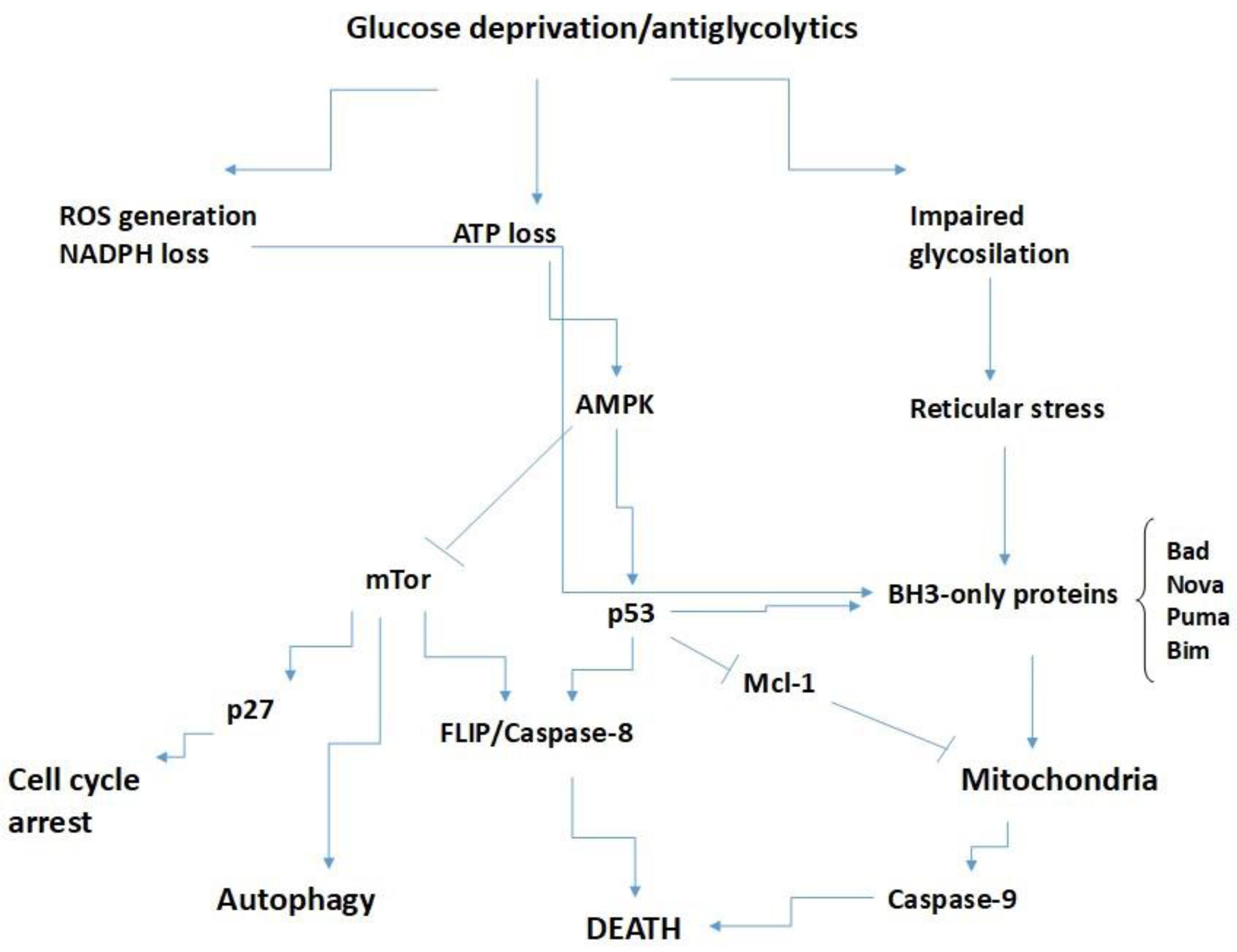
Adaptation of Cells to a Lack of Glucose
Glucose Controls BH3-Only Proteins
1.2.2. Possible Significance of Sugar Transporters in Cancer and Its Connection with Anticancer Therapy
1.2.3. Significance of Low-Carbohydrate Diets and Fasting
1.2.4. Targeting the Warburg Effect for Cancer Treatment: Ketogenic Diets for Management of Glioma
1.2.5. Dietary Changes for Effective Cancer Therapy
2. Conclusions
Final Conclusion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- González, D.; Bejarano, I.; Barriga, C.; Rodríguez, A.B.; Pariente, J.A. Oxidative Stress-Induced Caspases are Regulated in Human Myeloid HL-60 Cells by Calcium Signal. Curr. Signal Transduct. Ther. 2010, 5, 181–186. [Google Scholar] [CrossRef]
- González, D.; Espino, J.; Bejarano, I.; López, J.J.; Rodríguez, A.B.; Pariente, J.A. Caspase-3 and -9 are activated in human myeloid HL-60 cells by calcium signal. Mol. Cell. Biochem. 2010, 333, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Fadeel, B.; Orrenius, S. Apoptosis: A basic biological phenomenon with wide-ranging implications in human disease. J. Intern. Med. 2005, 258, 479–517. [Google Scholar] [CrossRef] [PubMed]
- Lawen, A. Apoptosis-an introduction. Bioessays 2003, 25, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Ashkenazi, A. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat. Rev. Cancer 2002, 2, 420–430. [Google Scholar] [CrossRef]
- Xu, G.; Shi, Y. Apoptosis signaling pathways and lymphocyte homeostasis. Cell Res. 2007, 17, 759–771. [Google Scholar] [CrossRef] [Green Version]
- Groenendyk, J.; Michalak, M. Endoplasmic reticulum quality control and apoptosis. Acta Biochim. Pol. 2005, 52, 381–395. [Google Scholar] [CrossRef]
- Bejarano, I.; Espino, J.; González-Flores, D.; Casado, J.G.; Redondo, P.C.; Rosado, J.A.; Barriga, C.; Pariente, J.A.; Rodríguez, A.B. Role of calcium signals on hydrogen peroxide-induced apoptosis in human myeloid HL-60 cells. Int. J. Biomed. Sci. 2009, 5, 246–256. [Google Scholar]
- Jones, P.A.; Baylin, S.B. The Epigenomics of Cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [Green Version]
- What Is Cancer?—National Cancer Institute. Available online: https://www.cancer.gov/about-cancer/understanding/what-is-cancer (accessed on 10 December 2021).
- Pereira, S.S.; Monteiro, M.P.; Antonini, S.R.; Pignatelli, D. Apoptosis regulation in adrenocortical carcinoma. Endocr. Connect. 2019, 8, R91–R104. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Arooj, S.; Wang, H. NK Cell-Based Immune Checkpoint Inhibition. Front. Immunol. 2020, 11, 167. [Google Scholar] [CrossRef]
- Li, X.B.; Gu, J.D.; Zhou, Q.H. Review of aerobic glycolysis and its key enzymes—New targets for lung cancer therapy. Thorac. Cancer 2015, 6, 17–24. [Google Scholar] [CrossRef]
- Shin, E.; Koo, J.S. Glucose Metabolism and Glucose Transporters in Breast Cancer. Front. Cell Dev. Biol. 2021, 9, 2404. [Google Scholar] [CrossRef]
- Wright, E.M. Glucose transport families SLC5 and SLC50. Mol. Asp. Med. 2013, 34, 183–196. [Google Scholar] [CrossRef]
- Aparicio, L.A.; Calvo, M.B.; Figueroa, A.; Pulido, E.G.; Campelo, R.G. Potential role of sugar transporters in cancer and their relationship with anticancer therapy. Int. J. Endocrinol. 2010, 2010, 205357. [Google Scholar] [CrossRef] [Green Version]
- Lizák, B.; Szarka, A.; Kim, Y.; Choi, K.S.; Németh, C.E.; Marcolongo, P.; Benedetti, A.; Bánhegyi, G.; Margittai, É. Glucose Transport and Transporters in the Endomembranes. Int. J. Mol. Sci. 2019, 20, 5898. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, I.M.; Aykin-Burns, N.; Sim, J.E.; Walsh, S.A.; Higashikubo, R.; Buettner, G.R.; Venkataraman, S.; Mackey, M.A.; Flanagan, S.W.; Oberley, L.W.; et al. Mitochondrial O2*- and H2O2 mediate glucose deprivation-induced stress in human cancer cells. J. Biol. Chem. 2005, 280, 4254–4263. [Google Scholar] [CrossRef] [Green Version]
- Aykin-Burns, N.; Ahmad, I.M.; Zhu, Y.; Oberley, L.W.; Spitz, D.R. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem. J. 2009, 418, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Yoon, J.H. Metabolic interplay between glycolysis and mitochondrial oxidation: The reverse Warburg effect and its therapeutic implication. World J. Biol. Chem. 2015, 6, 148. [Google Scholar] [CrossRef]
- Salazar, G. NADPH Oxidases and Mitochondria in Vascular Senescence. Int. J. Mol. Sci. 2018, 19, 1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Wahab, A.F.; Mahmoud, W.; Al-Harizy, R.M. Targeting glucose metabolism to suppress cancer progression: Prospective of anti-glycolytic cancer therapy. Pharmacol. Res. 2019, 150, 104511. [Google Scholar] [CrossRef]
- Ghanavat, M.; Shahrouzian, M.; Deris Zayeri, Z.; Banihashemi, S.; Kazemi, S.M.; Saki, N. Digging deeper through glucose metabolism and its regulators in cancer and metastasis. Life Sci. 2021, 264, 118603. [Google Scholar] [CrossRef] [PubMed]
- Tasevska, N.; Jiao, L.; Cross, A.J.; Kipnis, V.; Subar, A.F.; Hollenbeck, A.; Schatzkin, A.; Potischman, N. Sugars in diet and risk of cancer in the NIH-AARP Diet and Health Study. Int. J. Cancer 2012, 130, 159–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Mjiyad, N.; Caro-Maldonado, A.; Ramírez-Peinado, S.; Mũoz-Pinedo, C. Sugar-free approaches to cancer cell killing. undefined 2011, 30, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Plas, D.R.; Rathmell, J.C.; Fox, C.J.; Harris, M.H.; Thompson, C.B. Growth factors can influence cell growth and survival through effects on glucose metabolism. Mol. Cell. Biol. 2001, 21, 5899–5912. [Google Scholar] [CrossRef] [Green Version]
- Makarem, N.; Bandera, E.V.; Lin, Y.; Jacques, P.F.; Hayes, R.B.; Parekh, N. Consumption of Sugars, Sugary Foods, and Sugary Beverages in Relation to Adiposity-Related Cancer Risk in the Framingham Offspring Cohort (1991–2013). Cancer Prev. Res. 2018, 11, 347–358. [Google Scholar] [CrossRef] [Green Version]
- Chazelas, E.; Srour, B.; Desmetz, E.; Kesse-Guyot, E.; Julia, C.; Deschamps, V.; Druesne-Pecollo, N.; Galan, P.; Hercberg, S.; Latino-Martel, P.; et al. Sugary drink consumption and risk of cancer: Results from NutriNet-Santé prospective cohort. BMJ 2019, 366, l2408. [Google Scholar] [CrossRef] [Green Version]
- Buzzai, M.; Bauer, D.E.; Jones, R.G.; DeBerardinis, R.J.; Hatzivassiliou, G.; Elstrom, R.L.; Thompson, C.B. The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid beta-oxidation. Oncogene 2005, 24, 4165–4173. [Google Scholar] [CrossRef] [Green Version]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Coloff, J.L.; Ferguson, E.C.; Jacobs, S.R.; Cui, K.; Rathmell, J.C. Glucose Metabolism Attenuates p53 and Puma-dependent Cell Death upon Growth Factor Deprivation. J. Biol. Chem. 2008, 283, 36344. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.; Cory, S. The Bcl-2 protein family: Arbitres of cell survival. Science 1998, 281, 1322–1326. [Google Scholar] [CrossRef]
- Kelekar, A.; Thompson, C.B. Bcl-2-family proteins: The role of the BH3 domain in apoptosis. Trends Cell Biol. 1998, 8, 324–330. [Google Scholar] [CrossRef]
- Letai, A.; Bassik, M.C.; Walensky, L.D.; Sorcinelli, M.D.; Weiler, S.; Korsmeyer, S.J. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002, 2, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Reed, J.C. Mitochondrial control of cell death. Nat. Med. 2000, 6, 513–519. [Google Scholar] [CrossRef]
- Korsmeyer, S.J.; Wei, M.C.; Saito, M.; Weiler, S.; Oh, K.J.; Schlesinger, P.H. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000, 7, 1166–1173. [Google Scholar] [CrossRef]
- Hockenbery, D.; Nuñez, G.; Milliman, C.; Schreiber, R.D.; Korsmeyer, S.J. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature 1990, 348, 334–336. [Google Scholar] [CrossRef]
- Krajewski, S.; Tanaka, S.; Takayama, S.; Schibler, M.J.; Fenton, W.; Reed, J.C. Investigation of the subcellular distribution of the bcl-2 oncoprotein: Residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. AACR 1993, 53, 4701–4714. [Google Scholar]
- Zhu, W.; Cowie, A.; Wasfy, G.W.; Penn, L.Z.; Leber, B.; Andrews, D.W. Bcl-2 mutants with restricted subcellular location reveal spatially distinct pathways for apoptosis in different cell types. EMBO J. 1996, 15, 4130. [Google Scholar] [CrossRef]
- Murphy, K.M.; Ranganathan, V.; Farnsworth, M.L.; Kavallaris, M.; Lock, R.B. Bcl-2 inhibits Bax translocation from cytosol to mitochondria during drug-induced apoptosis of human tumor cells. Cell Death Differ. 2000, 7, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Mancinelli, F.; Caraglia, M.; Budillon, A.; Abbruzzese, A.; Bismuto, E. Molecular dynamics simulation and automated docking of the pro-apoptotic Bax protein and its complex with a peptide designed from the Bax-binding domain of anti-apoptotic Ku70. J. Cell. Biochem. 2006, 99, 305–318. [Google Scholar] [CrossRef]
- Nomura, M.; Shimizu, S.; Sugiyama, T.; Narita, M.; Ito, T.; Matsuda, H.; Tsujimoto, Y. 14-3-3 Interacts Directly with and Negatively Regulates Pro-apoptotic Bax. J. Biol. Chem. 2003, 278, 2058–2065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, H.Y.; Lavu, S.; Bitterman, K.J.; Hekking, B.; Imahiyerobo, T.A.; Miller, C.; Frye, R.; Ploegh, H.; Kessler, B.M.; Sinclair, D.A. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol. Cell 2004, 13, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Tsuruta, F.; Sunayama, J.; Mori, Y.; Hattori, S.; Shimizu, S.; Tsujimoto, Y.; Yoshioka, K.; Masuyama, N.; Gotoh, Y. JNK promotes Bax translocation to mitochondria through phosphorylation of 14-3-3 proteins. EMBO J. 2004, 23, 1889–1899. [Google Scholar] [CrossRef] [Green Version]
- Ghibelli, L.; Diederich, M. Multistep and multitask Bax activation. Mitochondrion 2010, 10, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Crompton, M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 1999, 341, 233. [Google Scholar] [CrossRef]
- Kuwana, T.; Newmeyer, D.D. Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr. Opin. Cell Biol. 2003, 15, 691–699. [Google Scholar] [CrossRef]
- Gillies, L.A.; Kuwana, T. Apoptosis regulation at the mitochondrial outer membrane. J. Cell. Biochem. 2014, 115, 632–640. [Google Scholar] [CrossRef]
- Kandasamy, K.; Srinivasula, S.; Alnemri, E.; Thompson, C.; Korsmeyer, S.; Bryant, J.; Srivastava, R.K. Involvement of proapoptotic molecules Bax and Bak in tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced mitochondrial disruption and apoptosis: Differential regulation of cytochrome c and Smac/DIABLO release. Cancer Res. 2003, 63, 1712–1721. [Google Scholar]
- Tan, K.O.; Fu, N.Y.; Sukumaran, S.K.; Chan, S.L.; Kang, J.H.; Poon, K.L.; Chen, B.S.; Yu, V.C. MAP-1 is a mitochondrial effector of Bax. Proc. Natl. Acad. Sci. USA 2005, 102, 14623–14628. [Google Scholar] [CrossRef] [Green Version]
- Lovell, J.F.; Billen, L.P.; Bindner, S.; Shamas-Din, A.; Fradin, C.; Leber, B.; Andrews, D.W. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 2008, 135, 1074–1084. [Google Scholar] [CrossRef] [Green Version]
- González-Flores, D.; De Nicola, M.; Bruni, E.; Caputo, F.; Rodríguez, A.B.; Pariente, J.A.; Ghibelli, L. Nanoceria protects from alterations in oxidative metabolism and calcium overloads induced by TNFα and cycloheximide in U937 cells: Pharmacological potential of nanoparticles. Mol. Cell. Biochem. 2014, 397, 245–253. [Google Scholar] [CrossRef]
- González-Flores, D.; Rodríguez, A.B.; Pariente, J.A. TNFα-induced apoptosis in human myeloid cell lines HL-60 and K562 is dependent of intracellular ROS generation. Mol. Cell. Biochem. 2014, 390, 281–287. [Google Scholar] [CrossRef]
- Scaffidi, C.; Fulda, S.; Srinivasan, A.; Friesen, C.; Li, F.; Tomaselli, K.J.; Debatin, K.M.; Krammer, P.H.; Peter, M.E. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 1998, 17, 1675–1687. [Google Scholar] [CrossRef] [Green Version]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Altman, B.J.; Coloff, J.L.; Herman, C.E.; Jacobs, S.R.; Wieman, H.L.; Wofford, J.A.; Dimascio, L.N.; Ilkayeva, O.; Kelekar, A.; et al. Glycogen synthase kinase 3alpha and 3beta mediate a glucose-sensitive antiapoptotic signaling pathway to stabilize Mcl-1. Mol. Cell. Biol. 2007, 27, 4328–4339. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.V.; Markowitz, S.; Zhou, S.; et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef] [Green Version]
- Haddad, M. The Impact of CB1 Receptor on Inflammation in Skeletal Muscle Cells. J. Inflamm. Res. 2021, 14, 3959, PMID 34421307; PMICID PMC8373309. [Google Scholar] [CrossRef]
- Haddad, M. The Impact of CB1 Receptor on Nuclear Receptors in Skeletal Muscle Cells. Pathophysiology 2021, 28, 457–470. [Google Scholar] [CrossRef]
- Haddad, M. Impact of Adenosine A2 Receptor Ligands on BCL2 Expression in Skeletal Muscle Cells. Appl. Sci. 2021, 11, 2272. [Google Scholar] [CrossRef]
- Sun, P.; Wang, H.; He, Z.; Chen, X.; Wu, Q.; Chen, W.; Sun, Z.; Weng, M.; Zhu, M.; Ma, D.; et al. Fasting inhibits colorectal cancer growth by reducing M2 polarization of tumor-associated macrophages. Oncotarget 2017, 8, 74649–74660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szypowska, A.; Regulska-Ilow, B. Significance of low-carbohydrate diets and fasting in patients with cancer. Rocz. Panstw. Zakl. Hig. 2019, 70, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Zuccoli, G.; Marcello, N.; Pisanello, A.; Servadei, F.; Vaccaro, S.; Mukherjee, P.; Seyfried, T.N. Metabolic management of glioblastoma multiforme using standard therapy together with a restricted ketogenic diet: Case Report. Nutr. Metab. 2010, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Rieger, J.; Bähr, O.; Maurer, G.D.; Hattingen, E.; Franz, K.; Brucker, D.; Walenta, S.; Kämmerer, U.; Coy, J.F.; Weller, M.; et al. ERGO: A pilot study of ketogenic diet in recurrent glioblastoma. Int. J. Oncol. 2014, 44, 1843–1852. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Longo, D.; Cook, L.S.; Weiss, N.S.; Potts, M.S.; Curtis, R.E.; Boice, J.D.; Hankey, B.F.; Fraumeni, J.F.; Machin, D.; West Andersen, K.; et al. Re: Breast Cancer Risk in Rats Fed a Diet High in n-6 Polyunsaturated Fatty Acids During Pregnancy. JNCI J. Natl. Cancer Inst. 1997, 89, 662–663. [Google Scholar] [CrossRef] [Green Version]
- Poff, A.; Koutnik, A.P.; Egan, K.M.; Sahebjam, S.; D’Agostino, D.; Kumar, N.B. Targeting the Warburg effect for cancer treatment: Ketogenic diets for management of glioma. Semin. Cancer Biol. 2019, 56, 135–148. [Google Scholar] [CrossRef]
- Kanarek, N.; Petrova, B.; Sabatini, D.M. Dietary modifications for enhanced cancer therapy. Nature 2020, 579, 507–517. [Google Scholar] [CrossRef]
- Brandhorst, S.; Choi, I.Y.; Wei, M.; Cheng, C.W.; Sedrakyan, S.; Navarrete, G.; Dubeau, L.; Yap, L.P.; Park, R.; Vinciguerra, M.; et al. A Periodic Diet that Mimics Fasting Promotes Multi-System Regeneration, Enhanced Cognitive Performance, and Healthspan. Cell Metab. 2015, 22, 86–99. [Google Scholar] [CrossRef] [Green Version]
- Nencioni, A.; Caffa, I.; Cortellino, S.; Longo, V.D. Fasting and cancer: Molecular mechanisms and clinical application. Nat. Rev. Cancer 2018, 18, 707–719. [Google Scholar] [CrossRef]
- Erickson, N.; Boscheri, A.; Linke, B.; Huebner, J. Systematic review: Isocaloric ketogenic dietary regimes for cancer patients. Med. Oncol. 2017, 34, 72. [Google Scholar] [CrossRef]
- Goncalves, M.D.; Lu, C.; Tutnauer, J.; Hartman, T.E.; Hwang, S.K.; Murphy, C.J.; Pauli, C.; Morris, R.; Taylor, S.; Bosch, K.; et al. High-fructose corn syrup enhances intestinal tumor growth in mice. Science 2019, 363, 1345–1349. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Patient | Diagnosis | Diet | After Treatment |
|---|---|---|---|
| 1 patient, 65 years old | Glioblastoma multiforme | 600 Kcal | The patient’s body mass reduced by roughly 20% after two months of administration of the diet and the presence of brain cancer cells was not observed. For six months, the stringent diet was continued. Relapse of the disease was identified by magnetic resonance imaging two months following the course of the same [65]. |
| 20 patients | Gliomas | Ketogenic diet (60 g of carbohydrates daily) that comprised, among other things, vegetable oils and fermented milk drinks (500 mL). | The median progression-free survival time was 5 weeks and the median survival was 32 weeks [64,66]. |
| 10 patients | End-stage cancer patients | Ketogenic diet for 28 days | The ketogenic diet may function in part by blocking insulin signaling in tumors [64]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalez-Flores, D.; Gripo, A.-A.; Rodríguez, A.-B.; Franco, L. Consequences of Glucose Enriched Diet on Oncologic Patients. Appl. Sci. 2023, 13, 2757. https://doi.org/10.3390/app13052757
Gonzalez-Flores D, Gripo A-A, Rodríguez A-B, Franco L. Consequences of Glucose Enriched Diet on Oncologic Patients. Applied Sciences. 2023; 13(5):2757. https://doi.org/10.3390/app13052757
Chicago/Turabian StyleGonzalez-Flores, David, Ana-Alejandra Gripo, Ana-Beatriz Rodríguez, and Lourdes Franco. 2023. "Consequences of Glucose Enriched Diet on Oncologic Patients" Applied Sciences 13, no. 5: 2757. https://doi.org/10.3390/app13052757
APA StyleGonzalez-Flores, D., Gripo, A.-A., Rodríguez, A.-B., & Franco, L. (2023). Consequences of Glucose Enriched Diet on Oncologic Patients. Applied Sciences, 13(5), 2757. https://doi.org/10.3390/app13052757