
Application of Quantum–Chemical Methods in the Forensic Prediction of Psychedelic Drugs’ Spectra (IR, NMR, UV–VIS, and MS): A Case Study of LSD and Its Analogs

Abstract
:Featured Application
Abstract
1. Introduction
2. Results and Discussion
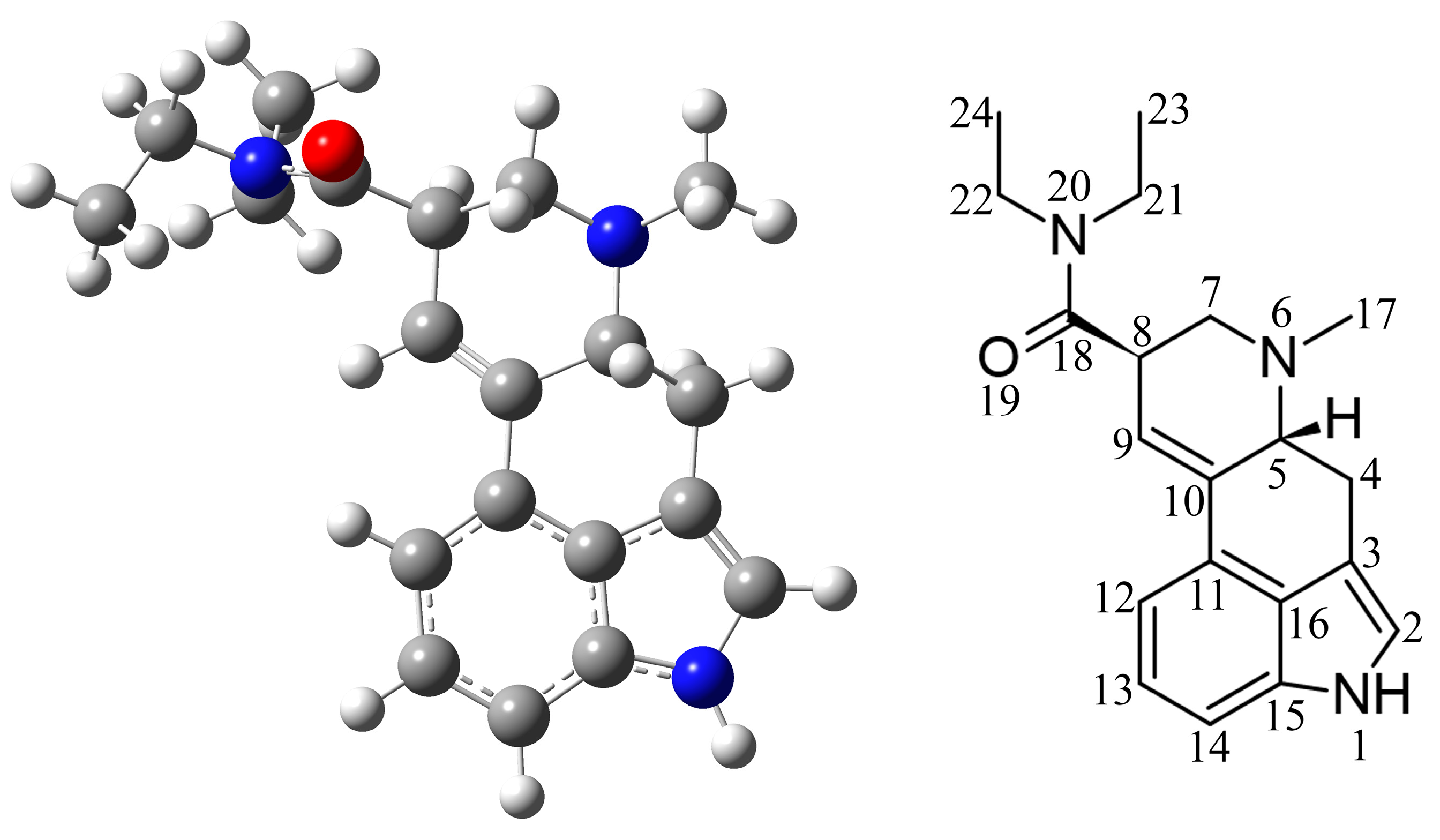
2.1. Selection of Appropriate Level of Theory
2.2. Experimental and Theoretical IR Spectra
2.3. Experimental and Theoretical NMR Spectra
2.4. Experimental and Theoretical UV–VIS Spectra
2.5. Experimental and Theoretical MS Spectra
2.6. Case Study of Real Samples Containing LSD
2.7. LSD Analogues, the Applicability of the Results
3. Materials and Methods
3.1. Spectra Acquisition
3.2. Theoretical Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- dos Santos, R.G.; Osório, F.L.; Crippa, J.A.S.; Hallak, J.E.C. Classical hallucinogens and neuroimaging: A systematic review of human studies. Neurosci. Biobehav. Rev. 2016, 71, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, R.G.; Osório, F.L.; Crippa, J.A.S.; Riba, J.; Zuardi, A.W.; Hallak, J.E.C. Antidepressive, anxiolytic, and antiaddictive effects of ayahuasca, psilocybin and lysergic acid diethylamide (LSD): A systematic review of clinical trials published in the last 25 years. Ther. Adv. Psychopharmacol. 2016, 6, 193–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libânio Osório Marta, R.F. Metabolism of lysergic acid diethylamide (LSD): An update. Drug Metab. Rev. 2019, 51, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Bouso, J.C.; dos Santos, R.G.; Alcázar-Córcoles, M.Á.; Hallak, J.E.C. Serotonergic psychedelics and personality: A systematic review of contemporary research. Neurosci. Biobehav. Rev. 2018, 87, 118–132. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, R.G.; Bouso, J.C.; Alcázar-Córcoles, M.Á.; Hallak, J.E.C. Efficacy, tolerability, and safety of serotonergic psychedelics for the management of mood, anxiety, and substance-use disorders: A systematic review of systematic reviews. Expert Rev. Clin. Pharmacol. 2018, 11, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Favretto, D.; Frison, G.; Maietti, S.; Ferrara, S.D. LC-ESI-MS/MS on an ion trap for the determination of LSD, iso-LSD, nor-LSD and 2-oxo-3-hydroxy-LSD in blood, urine and vitreous humor. Int. J. Legal Med. 2007, 121, 259–265. [Google Scholar] [CrossRef]
- da Cunha, K.F.; Kahl, J.M.M.; Fiorentin, T.R.; Oliveira, K.D.; Costa, J.L. High-sensitivity method for the determination of LSD and 2-oxo-3-hydroxy-LSD in oral fluid by liquid chromatography-tandem mass spectrometry. Forensic Toxicol. 2022, 40, 322–331. [Google Scholar] [CrossRef]
- Brandt, S.D.; Kavanagh, P.V.; Westphal, F.; Stratford, A.; Elliott, S.P.; Hoang, K.; Wallach, J.; Halberstadt, A.L. Return of the lysergamides. Part I: Analytical and behavioural characterization of 1-propionyl- d -lysergic acid diethylamide (1P-LSD). Drug Test. Anal. 2016, 8, 891–902. [Google Scholar] [CrossRef] [Green Version]
- Brandt, S.D.; Kavanagh, P.V.; Westphal, F.; Elliott, S.P.; Wallach, J.; Colestock, T.; Burrow, T.E.; Chapman, S.J.; Stratford, A.; Nichols, D.E.; et al. Return of the lysergamides. Part II: Analytical and behavioural characterization of N 6 -allyl-6-norlysergic acid diethylamide (AL-LAD) and (2’ S,4’ S )-lysergic acid 2,4-dimethylazetidide (LSZ). Drug Test. Anal. 2017, 9, 38–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandt, S.D.; Kavanagh, P.V.; Westphal, F.; Elliott, S.P.; Wallach, J.; Stratford, A.; Nichols, D.E.; Halberstadt, A.L. Return of the lysergamides. Part III: Analytical characterization of N 6 -ethyl-6-norlysergic acid diethylamide (ETH-LAD) and 1-propionyl ETH-LAD (1P-ETH-LAD). Drug Test. Anal. 2017, 9, 1641–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandt, S.D.; Kavanagh, P.V.; Twamley, B.; Westphal, F.; Elliott, S.P.; Wallach, J.; Stratford, A.; Klein, L.M.; McCorvy, J.D.; Nichols, D.E.; et al. Return of the lysergamides. Part IV: Analytical and pharmacological characterization of lysergic acid morpholide (LSM-775). Drug Test. Anal. 2018, 10, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Halberstadt, A.L.; Klein, L.M.; Chatha, M.; Valenzuela, L.B.; Stratford, A.; Wallach, J.; Nichols, D.E.; Brandt, S.D. Pharmacological characterization of the LSD analog N-ethyl-N-cyclopropyl lysergamide (ECPLA). Psychopharmacology 2019, 236, 799–808. [Google Scholar] [CrossRef] [Green Version]
- Nichols, D.E. Hallucinogens. Pharmacol. Ther. 2004, 101, 131–181. [Google Scholar] [CrossRef]
- Grumann, C.; Henkel, K.; Stratford, A.; Hermanns-Clausen, M.; Passie, T.; Brandt, S.D.; Auwärter, V. Validation of an LC-MS/MS method for the quantitative analysis of 1P-LSD and its tentative metabolite LSD in fortified urine and serum samples including stability tests for 1P-LSD under different storage conditions. J. Pharm. Biomed. Anal. 2019, 174, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Spálovská, D.; Maříková, T.; Kohout, M.; Králík, F.; Kuchař, M.; Setnička, V. Methylone and pentylone: Structural analysis of new psychoactive substances. Forensic Toxicol. 2019, 37, 366–377. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Cañamares, M.V.; Kakoulli, I.; Sanchez-Cortes, S. Chemical Characterization and Molecular Dynamics Simulations of Bufotenine by Surface-Enhanced Raman Scattering (SERS) and Density Functional Theory (DFT). J. Phys. Chem. Lett. 2022, 13, 5831–5837. [Google Scholar] [CrossRef] [PubMed]
- Hosseinian, A.; Vessally, E.; Bekhradnia, A.; Nejati, K.; Rahimpour, G. Benzoylethanamine drug interaction with the AlN nanosheet, nanotube and nanocage: Density functional theory studies. Thin Solid Film. 2017, 640, 93–98. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. IUCr The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Baker, R.W.; Chothia, C.; Pauling, P.; Weber, H.P. Molecular Structures of Hallucinogenic Substances: Lysergic Acid Diethylamide, Psilocybin, and 2,4,5-Trimethoxyamphetamine. Mol. Pharmacol. 1973, 9, 23–32. [Google Scholar] [PubMed]
- Baker, R.W.; Chothia, C.; Pauling, P.; Weber, H.P. Molecular Structure of LSD. Science 1972, 178, 614–615. [Google Scholar] [CrossRef] [PubMed]
- Shobana, D.; Sudha, S.; Ramarajan, D.; Dimić, D. Synthesis, crystal structure, spectral characterization and Hirshfeld surface analysis of (E)-N′-(3-ethoxy-4-hydroxybenzylidene)-4-fluorobenzohydrazide single-crystal—A novel NLO active material. J. Mol. Struct. 2022, 1250, 131856. [Google Scholar] [CrossRef]
- Dennington, R.; Todd, K.; Millam, J. Gauss View; Semichem Inc.: Shawnee, KS, USA, 2009. [Google Scholar]
- Jamroz, M.H. Vibrational Energy Distribution Analysis VEDA 4, Warsaw, 2004–2010. Available online: https://smmg.pl/software/veda (accessed on 1 December 2022).
- Mesley, R.J.; Evans, W.H. Infrared identification of lysergide (LSD). J. Pharm. Pharmacol. 2011, 21, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Dimić, D.S.; Kaluđerović, G.N.; Avdović, E.H.; Milenković, D.A.; Živanović, M.N.; Potočňák, I.; Samoľová, E.; Dimitrijević, M.S.; Saso, L.; Marković, Z.S.; et al. Synthesis, Crystallographic, Quantum Chemical, Antitumor, and Molecular Docking/Dynamic Studies of 4-Hydroxycoumarin-Neurotransmitter Derivatives. Int. J. Mol. Sci. 2022, 23, 1001. [Google Scholar] [CrossRef] [PubMed]
- Dimić, D.; Milenković, D.; Ilić, J.; Šmit, B.; Amić, A.; Marković, Z.; Dimitrić Marković, J. Experimental and theoretical elucidation of structural and antioxidant properties of vanillylmandelic acid and its carboxylate anion. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 198, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Dimić, D.; Milenković, D.; Marković, Z.; Marković, J.D. Structural and spectral analysis of 3-metoxytyramine, an important metabolite of dopamine. J. Mol. Struct. 2017, 1134, 226–236. [Google Scholar] [CrossRef]
- Avdović, E.H.; Dimić, D.S.; Dimitrić Marković, J.M.; Vuković, N.; Radulović, M.Đ.; Živanović, M.N.; Filipović, N.D.; Đorović, J.R.; Trifunović, S.R.; Marković, Z.S. Spectroscopic and theoretical investigation of the potential anti-tumor and anti-microbial agent, 3-(1-((2-hydroxyphenyl)amino)ethylidene)chroman-2,4-dione. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 206, 421–429. [Google Scholar] [CrossRef]
- Brandt, S.D.; Kavanagh, P.V.; Westphal, F.; Stratford, A.; Elliott, S.P.; Dowling, G.; Wallach, J.; Halberstadt, A.L. Return of the lysergamides. Part V: Analytical and behavioural characterization of 1-butanoyl-d-lysergic acid diethylamide (1B-LSD). Drug Test. Anal. 2019, 11, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Dimić, D. The importance of specific solvent–solute interactions for studying UV–VIS spectra of light-responsive molecular switches. Comptes Rendus Chim. 2018, 21, 1001–1010. [Google Scholar] [CrossRef]
- Bellman, S.W. Mass Spectral Identification of Some Hallucinogenic Drugs. J. AOAC Int. 1968, 51, 164–175. [Google Scholar] [CrossRef]
- Clark, C.C. The Differentiation of Lysergic Acid Diethylamide (LSD) from N -Methyl- N -Propyl and N -Butyl Amides of Lysergic Acid. J. Forensic Sci. 1989, 34, 12674J. [Google Scholar] [CrossRef]
- Nakahara, Y.; Niwaguchi, T. Studies on Lysergic Acid Diethylamide and Related Compounds. I. Synthesis of d-N6-Demethyl-lysergic Acid Diethylamide. Chem. Pharm. Bull. 1971, 19, 2337–2341. [Google Scholar] [CrossRef] [Green Version]
- Shobana, D.; Sudha, S.; Ramarajan, D.; Ristivojević, N.; Rakić, A.; Dimić, D. Structural, spectroscopic (IR, Raman, and NMR), quantum chemical, and molecular docking analysis of (E)-2-(2,5-dimethoxybenzylidene)hydrazinecarbothioamide and its dimers. J. Mol. Struct. 2022, 1247, 131277. [Google Scholar] [CrossRef]
- Monte, A.P.; Marona-Lewicka, D.; Kanthasamy, A.; Sanders-Bush, E.; Nichols, D.E. Stereoselective LSD-like Activity in a Series of d-Lysergic Acid Amides of (R)- and (S)-2-Aminoalkanes. J. Med. Chem. 1995, 38, 958–966. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2006, 2, 364–382. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B-Condens. Matter Mater. Phys. 1996, 54, 16533–16539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Munos, R.A.; Panchenko, Y.N.; Koptev, G.S.; Stepanov, N.F. Program for calculating distribution of potential energy in internal coordinates. J. Appl. Spectrosc. 1970, 12, 428–429. [Google Scholar] [CrossRef]
- Jacquemin, D.; Preat, J.; Perpète, E.A. A TD-DFT study of the absorption spectra of fast dye salts. Chem. Phys. Lett. 2005, 410, 254–259. [Google Scholar] [CrossRef]
- Jacquemin, D.; Perpète, E.A. Ab initio calculations of the colour of closed-ring diarylethenes: TD-DFT estimates for molecular switches. Chem. Phys. Lett. 2006, 429, 147–152. [Google Scholar] [CrossRef]
- Zieliński, R.; Szymusiak, H. Application of Dft B3Lyp/Giao and B3Lyp/Csgt Methods for Interpretation of Nmr Spectra of Flavonoids. Pol. J. Food Nutr. Sci. 2003, 53, 157–162. [Google Scholar]
- Bohmann, J.A.; Weinhold, F.; Farrar, T.C. Natural chemical shielding analysis of nuclear magnetic resonance shielding tensors from gauge-including atomic orbital calculations. J. Chem. Phys. 1997, 107, 1173. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| B3LYP | B3LYP-D3BJ | CAM-B3LYP | B3PW91 | M05-2X | M06-2X | |
|---|---|---|---|---|---|---|
| Bond lengths (Å) | 0.0423 | 0.0430 | 0.0444 | 0.0439 | 0.0436 | 0.0437 |
| Bond angles (°) | 2.88 | 2.82 | 2.83 | 2.85 | 2.70 | 2.72 |
| Carbon Atom | Experimental (ppm) | Calculated, Unscaled (ppm) | Calculated, Scaled (ppm) |
|---|---|---|---|
| C24 | 13.1 | 13.5 | 11.4 |
| C22 | 14.8 | 15.5 | 13.2 |
| C4 | 27.3 | 32.8 | 27.8 |
| C23 | 39.9 | 44.7 | 37.9 |
| C21 | 40.2 | 46.0 | 39.0 |
| C17 | 42.0 | 46.8 | 39.7 |
| C8 | 43.9 | 48.5 | 41.1 |
| C7 | 56.0 | 60.8 | 51.5 |
| C5 | 63.2 | 71.2 | 60.3 |
| C14 | 109.7 | 130.9 | 110.9 |
| C3 | 110.8 | 132.6 | 112.4 |
| C12 | 112.6 | 133.4 | 113.2 |
| C2 | 118.3 | 139.3 | 118.0 |
| C9 | 119.7 | 146.2 | 123.9 |
| C13 | 123.3 | 146.4 | 124.1 |
| C16 | 126.3 | 149.0 | 126.3 |
| C11 | 128.1 | 152.4 | 129.2 |
| C15 | 134.0 | 155.4 | 131.8 |
| C10 | 136.3 | 166.3 | 141.0 |
| C18 | 170.9 | 196.4 | 166.5 |
| R | 0.999 | 0.999 | |
| MAE (ppm) | 14.9 | 2.0 | |
| Experimental [nm] | CPCM [nm] | CPCM + 1 Molecule Water (nm) | Excited State Optimization (nm) |
|---|---|---|---|
| 314 | 288 (HOMO→LUMO, 96%, f = 0.3948) | 291 (HOMO→LUMO, 96%, f = 0.3793) | 330 (HOMO→LUMO) |
| 222 | 225 (HOMO→LUMO+1, 32%, f = 0.2307) | 226 (HOMO→LUMO+2, 37%, f = 0.1816) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Džodić, J.; Milenković, D.; Marković, M.; Marković, Z.; Dimić, D. Application of Quantum–Chemical Methods in the Forensic Prediction of Psychedelic Drugs’ Spectra (IR, NMR, UV–VIS, and MS): A Case Study of LSD and Its Analogs. Appl. Sci. 2023, 13, 2984. https://doi.org/10.3390/app13052984
Džodić J, Milenković D, Marković M, Marković Z, Dimić D. Application of Quantum–Chemical Methods in the Forensic Prediction of Psychedelic Drugs’ Spectra (IR, NMR, UV–VIS, and MS): A Case Study of LSD and Its Analogs. Applied Sciences. 2023; 13(5):2984. https://doi.org/10.3390/app13052984
Chicago/Turabian StyleDžodić, Jelica, Dejan Milenković, Milica Marković, Zoran Marković, and Dušan Dimić. 2023. "Application of Quantum–Chemical Methods in the Forensic Prediction of Psychedelic Drugs’ Spectra (IR, NMR, UV–VIS, and MS): A Case Study of LSD and Its Analogs" Applied Sciences 13, no. 5: 2984. https://doi.org/10.3390/app13052984