1. Introduction
One of the tasks of molecular photonics is the development of electroluminescent devices for emitting information panels, optoelectronic devices, etc. A possible polymeric material for solving this problem can be both photoactive, electrically conductive matrices and polymeric ligands. In addition, an important approach is the use of thin electroluminescent polymer layers instead of materials based on low molecular weight phosphors. The advantage of this approach is the rejection of energy-consuming vacuum deposition of phosphors in favor of solution technologies compatible with the deposition of coatings in a centrifugal field and with the use of inkjet, screen, and matrix printing technologies for optoelectronic circuits [
1,
2].
In the arsenal of polymer physics and chemistry, there are several ways to create new materials for use in electroluminescent devices. These include, first, the synthesis of highly efficient electroluminescent homo- and copolymers [
3,
4]; secondly, the creation of composite materials (for example, the dispersion of luminescent quantum dots in a conductive polymer matrix) [
5,
6,
7,
8]. In addition, the design of organo−inorganic hybrid photo- and electroluminescent materials, for example, based on metal−polymer complexes (MPCs), is also an interesting direction [
9,
10,
11,
12,
13].
In the latter case, lanthanide ions Ln
3+ are often used as the luminescent component, which is due to the special structure of the outer electronic levels of three-charged rare-earth ions: the energies of these levels are well separated due to screening of 4f-orbitals by electrons of 5s- and 5p-sublevels; intra-level transitions 4f–4f, covering the region of visible and near infrared radiation, are clear and well recognized as well. Thus, the luminescence of Ln
3+ ions is characterized by quasi-monochromatic emission (the half-width of the luminescence bands is 5–10 nm, while for organic chromophores it is up to 100 nm), the stability of emission over time, the independence of the position of the luminescence bands from the nature of the ligand and solvent, and a large pseudo-Stokes shift in complexes, as well as a long lifetime of the excited state (≈1000 μs) [
14,
15].
The main problem is the selection and synthesis of a polymeric ligand capable of providing efficient intramolecular transfer of electronic excitation energy to the central Ln3+ atom. Units not participating in complexation should provide the desired optical and electrically conductive properties.
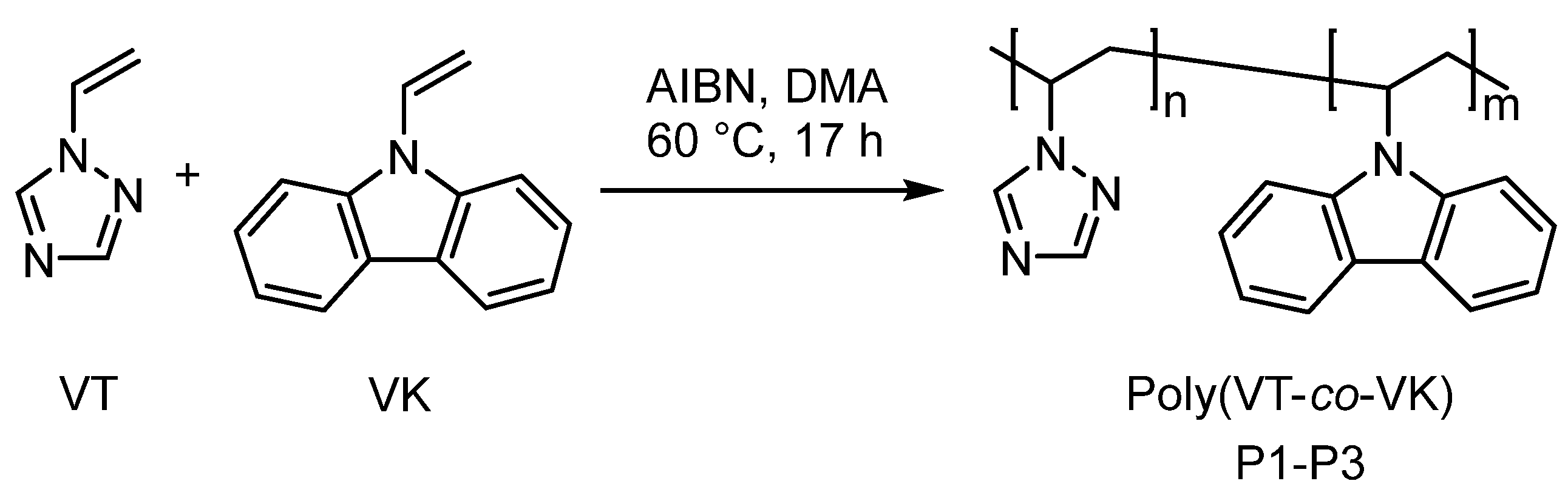
To develop a polymer ligand with unique properties, we chose promising monomers, namely N-vinylcarbazole (VK) and 1-vinyl-1,2,4-triazole (VT), for the synthesis of new effective copolymers. This choice was because the synthetic features, physicochemical, and optical properties of homopolymers based on VK and VT have been thoroughly studied, widely presented in scientific publications, and testify to their originality.
It has been established that poly(
N-vinylcarbazole) (PVK) is characterized by mechanical strength, high chemical and thermal stability, and has a predominantly
p-type electrical conductivity [
16]. PVK exhibits a high photoelectric sensitivity in the UV region of the spectrum, which increases significantly upon sensitization of the polymer with various dopants (di-, tri-, and tetranitro-9-fluorenone;
p-chloranil; and
p-bromanil), while the maximum of the spectral dependence of the photosensitivity shifts to the visible region of the spectrum [
17,
18,
19]. On the basis of PVK, effective electrophotographic layers have been developed that are used to develop information carriers. PVK is promising for the development of photosensitive layers of solar cells, organic light-emitting diodes (OLED), and organic field effect transistors (OFET) [
20,
21].
It has been established that homo- and copolymers based on 1-vinyl-1,2,4-triazole have a wide range of valuable properties, such as good solubility in water and polar organic solvents, chemical resistance and thermal stability, biocompatibility, and non-toxicity, as well as the ability of complex formation [
22,
23,
24]. 1-Vinyl-1,2,4-triazole under conditions of controlled radical polymerization with reversible chain transfer by the addition-fragmentation (RAFT) mechanism can form narrowly dispersed polymers with molecular weights ranging from 11 to 61 kDa [
25]. Using RAFT polymerization, amphiphilic block copolymers of various compositions containing successive block fragments of triazole and carbazole units have been obtained [
18].
Polymers based on 1-vinyl-1,2,4-triazole exhibit a high stabilizing ability in the formation of polymer nanocomposites with nanoparticles of various metals (Ag, Au, Fe, and so on) [
22,
26,
27]. This is because of the presence of pyridine nitrogen atoms in the triazole fragment of macromolecules, which are capable of efficiently entering into coordination interactions with metal ions. Synthesized nanocomposites with silver nanoparticles have a high antibacterial activity against Gram-positive and Gram-negative microorganisms [
28,
29]. The ability of PVT for complex formation determined its high sorption capacity regarding Pd(II) and Pt(IV) ions [
30].
Because of their film-forming ability, as well as electrochemical, mechanical, and thermal stability (up to 300–330 °C), VT (co)polymers are promising for the development of proton-conducting fuel cell membranes that are capable of providing their high ionic conductivity (up to 10
−3–10
−1 S/cm) under anhydrous conditions at temperatures above 100 °C [
31].
Thus, taking into account the original properties of 1-vinyl-1,2,4-triazole and N-vinylcarbazole monomers, we synthesized new copolymers based on them by radical copolymerization and obtained copolymers whose macromolecules involve hole-conducting leading carbazole fragments and triazole units with complexing and plastic properties. By varying the ratio of starting monomers, copolymers of different compositions were obtained. So, the formation of polymeric luminescent systems was performed in three main stages. In the first stage, functional triazole−carbazole copolymers, poly(VT-co-VK), of different compositions were synthesized by free-radical copolymerization. Next, a polymer complex with terbium (III) ions (Tb3+) was obtained by donor−acceptor binding. In the last stage, the MPC was introduced into an inert matrix of polymethylmethacrylate (PMMA) in order to obtain a luminescent organic−inorganic composite. The processes of the formation of metal−polymer complexes based on synthesized polymer ligands with Tb3+, which can provide the material with the desired luminescent properties in the visible range (green glow), were studied. The photophysical properties of the obtained MPCs were studied, making it possible to assess the prospects and practical significance of new materials.
2. Materials and Methods
Materials
1-Vinyl-1,2,4-triazole was synthesized and characterized by the procedure described in [
20].
N-Vinylcarbazole (Sigma-Aldrich, St. Louis, MO, USA) and azobisisobutyronitrile (AIBN, Merck, Rahway, NJ, USA) were used as received. Dimethylacetamide (DMA), acetone, and ethanol were distilled under reduced pressure and were purified before use according to known procedures. Other materials were used without further purification.
Synthesis of poly(1-vinyl-1,2,4-triazole-
co-N-vinylcarbazole) (poly(VT–
co–VK))–general procedure: Poly(VT–
co–VK) was synthesized by radical copolymerization of VT and VK monomers using AIBN as a radical initiator. Copolymerization was performed in a sealed glass ampule in solution DMA at 60 °C for 17 h under an argon atmosphere. After the reaction was complete, the glass ampule was left to cool to room temperature and opened. The reaction mixture was diluted with DMA and the solution was precipitated twice into an acetone−ethanol mixture. The precipitate formed was centrifuged at 5000 rpm for 10 min and dried in vacuo at 50 °C for 24 h. The copolymers were white powders. By varying the ratio of VT and VK monomers in the initial reaction mixture from 93:7, 85:15, and 70:30 mol. % (
Table 1), three different compositions of copolymers were obtained. The yield was 91–94%.
Synthesis of poly(VT–co–VK) P1: The reaction mixture: VT (0.87 g, 3 mol/L), VK (0.13 g, 43 mol/L), AIBN (17.7 mg, 0.03 mol/L), DMA (3.06 g). Yield: 0.94 g, 94%. FT-IR: ν = 747 cm−1 (−CH=CH2), 1222 cm−1 (C=CH2), 1334 cm−1 (C–N), 1452 cm−1 (-CH), 1485 cm−1 (–C=C–) for VK; 661 cm−1 (C–N), 1004 cm−1 (C–H), 1140 (C–H), 1276 cm−1 (N–N), 1434 cm−1 (C–N), 1505 cm−1 (C=N) for triazole ring; 2934 cm−1 (CH2), 3110 cm−1 (C–H) for polymer chain. 1H NMR (400 MHz, DMSO-d6): δ = 1.40–2.25 (br, 2H, CH2 in the polymer main chain), 2.6 –4.15 (br, 1H, >CH– in the polymer main chain), 7.50–8.15 ppm (br, 2H, triazole ring), 7.50–8.15 ppm (br, 8H, carbazole ring).
Synthesis of poly(VT–co–VK) P2: The reaction mixture: VT (0.74 g, 3 mol/L), VK (0.26 g, 43 mol/L), AIBN (17.7 mg, 0.03 mol/L), DMA (2.80 g). Yield: 0.93 g, 93%. FT-IR: ν = 745 cm−1 (−CH=CH2), 1222 cm−1 (C=CH2), 1334 cm−1 (C–N), 1452 cm−1 (-CH), 1483 cm−1 (–C=C–) for VK; 661 cm−1 (C–N), 1003 cm−1 (C–H), 1140 (C–H), 1276 cm−1 (N–N), 1434 cm−1 (C–N), 1504 cm−1 (C=N) for triazole ring; 2933 cm−1 (CH2), 3112 cm−1 (C–H) for polymer chain. 1H NMR (400 MHz, DMSO-d6): δ = 1.40–2.25 (br, 2H, CH2 in the polymer main chain), 2.6 –4.15 (br, 1H, >CH– in the polymer main chain), 7.50–8.15 ppm (br, 2H, triazole ring), 7.50–8.15 ppm (br, 8H, carbazole ring).
Synthesis of poly(VT–co–VK) P3: The reaction mixture: VT (0.54 g, 3 mol/L), VK (0.47 g, 43 mol/L), AIBN (17.7 mg, 0.03 mol/L), DMA (2.34 g). Yield: 0.91 g, 91%. FT-IR: ν = 744 cm−1 (−CH=CH2), 1224 cm−1 (C=CH2), 1333 cm−1 (C–N), 1452 cm−1 (-CH), 1483 cm−1 (–C=C–) for VK; 662 cm−1 (C–N), 1003 cm−1 (C–H), 1139 (C–H), 1275 cm−1 (N–N), 1434 cm−1 (C–N), 1503 cm−1 (C=N) for triazole ring; 2932 cm−1 (CH2), 3114 cm−1 (C–H) for polymer chain. 1H NMR (400 MHz, DMSO-d6): δ = 1.40–2.25 (br, 2H, CH2 in the polymer main chain), 2.6 –4.15 (br, 1H, >CH– in the polymer main chain), 7.50–8.15 ppm (br, 2H, triazole ring), 7.50–8.15 ppm (br, 8H, carbazole ring).
Instrumentation
1H NMR (400.13 MHz) spectra were recorded with a Bruker DPX-400 spectrometer at room temperature. The FT-IR spectra were recorded using a Varian 3100 FTIR spectrometer (Palo Alto, Santa Clara, CA, USA). The number-average molecular weight (Mn), weight-average molecular weight (Mw), and molecular weight distribution (Mw/Mn) were determined by size-exclusion chromatography using a Shimadzu LC-20 Prominence system (Shimadzu Corporation, Kyoto, Japan), equipped with a differential refractive index detector Shimadzu RID-20A at 50 °C. The column was Agilent PolyPore 7.5 × 300 mm (PL1113-6500) with a guard column. The system was operated at the flow rate of 0.7 mL/min using DMF containing 0.05M LiBr as the eluent. A set of 12 polystyrene standards (Polystyrene High EasiVials PL2010-0201, Agilent, Santa Clara, CA, USA) ranging from 162 to 6,570,000 g/mol was employed for calibration. STA 449 Jupiter (Netzsch, Selb, Germany) was used to perform the thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) in the air at a heating rate of 5 °C/min from 20 to 800 °C. The copolymers had an electrical conductivity of 10−13 S/cm; after doping with iodine for 48 h, the electrical conductivity increased by four orders of magnitude (10−9 S/cm).
Metal−polymer complexes of the synthesized VT–VK polymer ligands and Tb3+ ions were prepared in DMF. Films of the studied MPC in a poly(methyl methacrylate) (PMMA) matrix were obtained as follows. To solutions containing 50 mg of PMMA in DMF, the previously prepared solutions of the obtained MPC were added to a total volume of 2 mL. The polymer concentration depended on the content of VK units, as the concentration of the latter was brought up to 10−3 M in all cases. The [Tb3+]:[VK] ratio was 1:1. The complex was obtained from the solution using the salt TbCl₃ x 6H₂O. The finally prepared solution was poured onto a glass substrate into rings with an area of 4 cm2 and dried at 60 °C to a constant weight. The mass concentration of Tb3+ ions (~0.63%) and VK units (~0.77%) in the resulting films was constant, which is necessary for a correct comparison of the luminescence intensities for MPC based on copolymers of different compositions.
Absorption spectra of poly(VT-co-VK) solutions were recorded on a spectrophotometer SF-256 UVI (LOMO, St. Petersburg, Russia) in the range from 250 to 450 nm. The polymer concentration in DMF was chosen in the range from 0.1 to 1 mg/mL. The photoluminescence spectra of both the synthesized VT–VK copolymers and their metal–polymer complexes with Tb3+ ions were recorded on an LS-100 BASE luminescence spectrophotometer (PTI® Lasers INC, London–Toronto, ON, Canada) equipped with a solid sample holder for polymer films. The luminescent signal was measured from the side of the incidence of the exciting beam. The spectral width of the slits at the input and output of the observation and excitation monochromators was 4 nm. Luminescence excitation at 300 nm was used. Photoluminescence in the fluorescent mode was observed in the range of 320–700 nm, covering almost the entire visible range. The magnitude of the luminescence intensity was numerically reduced to a single PMT gain, taking into account the laboratory standard. Such a reduction in the measured luminescence signal made it possible to compare the relative intensity for different conditions, taking into account the fact that in this work, the concentration of Tb3+ ions and VK units was maintained constantly.
3. Results and Discussion
Free radical copolymerization of 1-vinyl-1,2,4-triazole with
N-vinylcarbazole was carried out at different comonomer ratios in a dimethylacetamide solution under the action of an azobisisobutyronitrile initiator in a sealed ampule in an argon atmosphere. Copolymerization proceeds at a temperature of 60 °C. The concentration of VT and VK comonomers in the solution and the concentration of the initiator were 3 M and 3 × 10
−2 M, respectively (
Scheme 1 and
Table 1).
The compositions of the copolymers were determined from the UV spectra measured at 344 nm (VK absorption band). The extinction coefficients for the comonomer and monomer were taken as equal to the extinction coefficient of the polyvinylcarbazole ε = 3500 L/(mol∙cm).
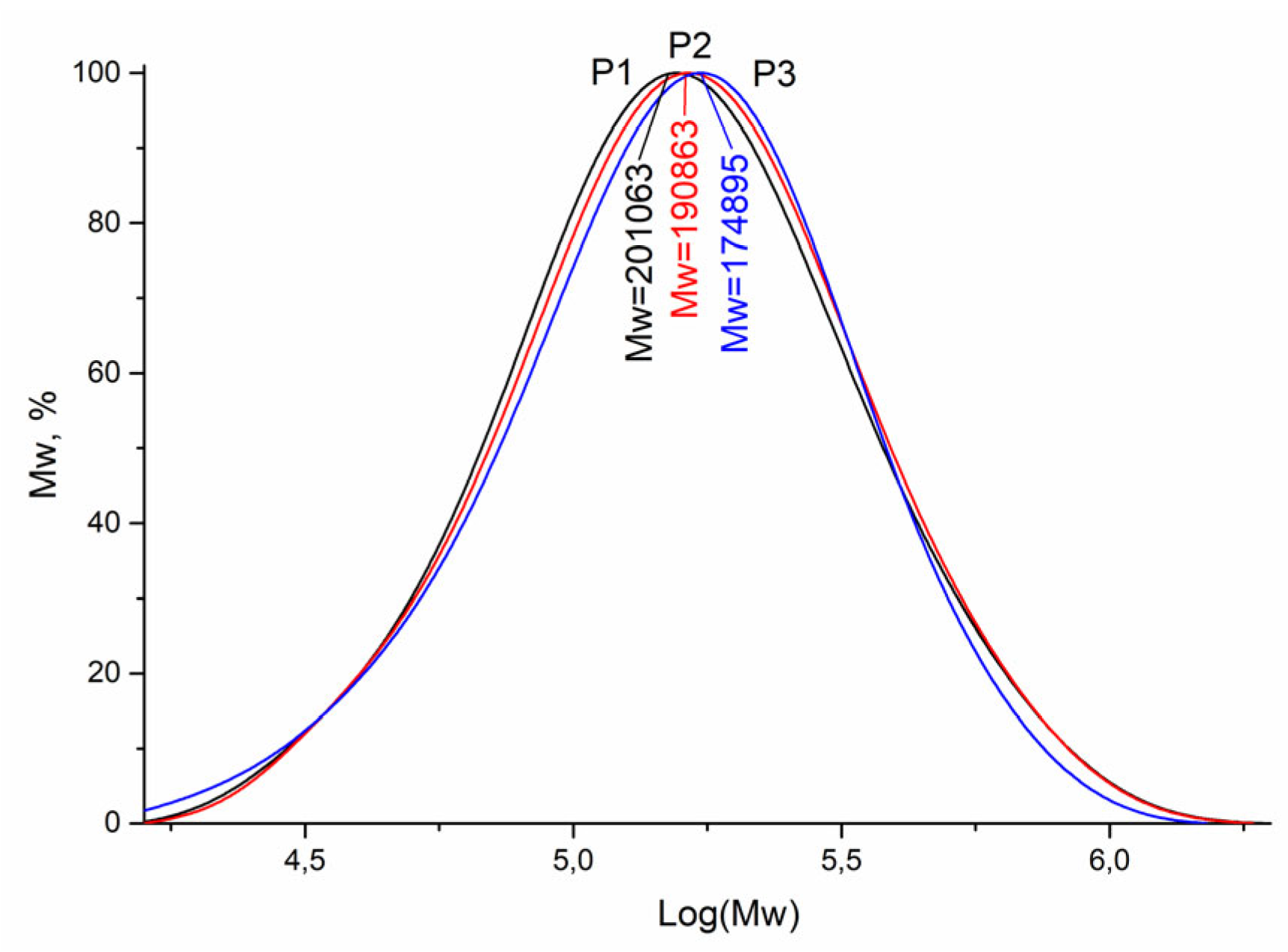
The molecular weight distribution of the obtained P1–P3 copolymers was studied using gel permeation chromatography (
Figure 1).
The molecular weight of P1–P3 copolymers depends on their composition. It should be noted that with an increase in the carbazole units in the copolymer and a decrease in the vinyltriazole units, the weight-average molecular weight decreased from 201 to 175 kDa upon passing from the P1 to P3 copolymer. The dispersity (
Đ) of the prepared P1–P3 copolymers was in the range of 1.76–1.79, i.e., the value of
Đ did not change significantly (
Table 1). Thus, the synthesized copolymers showed a unimodal molecular weight distribution.
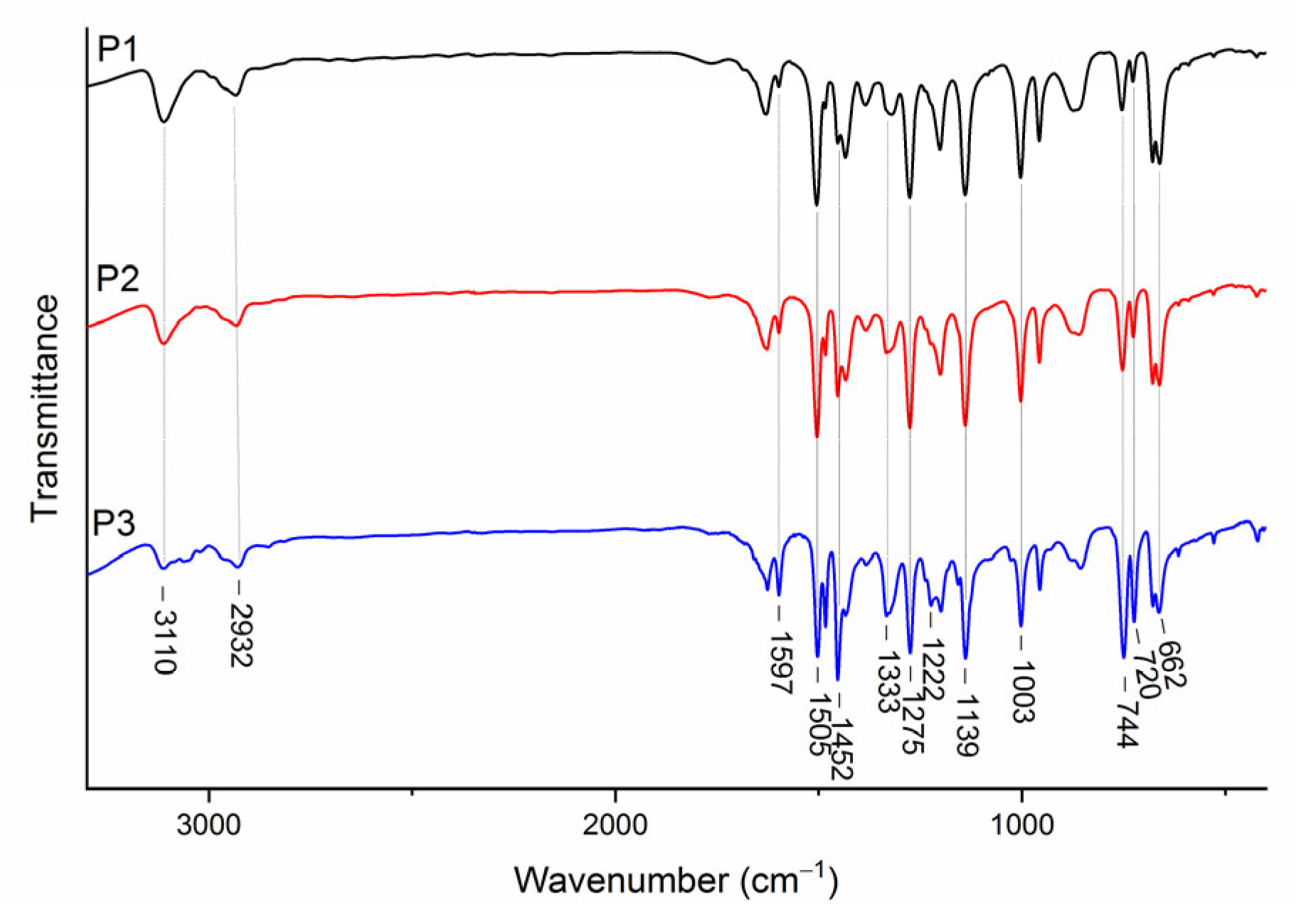
The FT-IR spectra of the synthesized P1–P3 copolymers are shown in
Figure 2. In the P1–P3 spectra, the absorption bands of stretching vibrations of the double bonds of the initial VT and VK monomers disappeared at 1640 cm
−1. This indicates the absence of the initial 1-vinyl-1,2,4-triazole and
N-vinylcarbazole comonomers in the copolymer.
The IR spectra of the P1–P3 copolymers show that the characteristic absorption bands of stretching and bending vibrations at 3110–3114 (C–H) and 2932–2934 cm−1 (CH2) were retained for the polymer chain. The IR spectra of P1–P3 poly(VT-co-VK) copolymers contained characteristic bands of stretching and bending vibrations of the triazole ring at 1503–1505 (C=N), 1434 (C–N), 1275–1276 (N–N), 1139–1140 (C–H), 1003–1004 (C–H), and 661–663 cm−1 (C–N). The presence of the carbazole units in the IR spectra of P1–P3 copolymers was confirmed by absorption bands at 1597, 1483–1485, 1452, 1333–1334, 1222–1224, 744–747, and 720–723 cm−1.
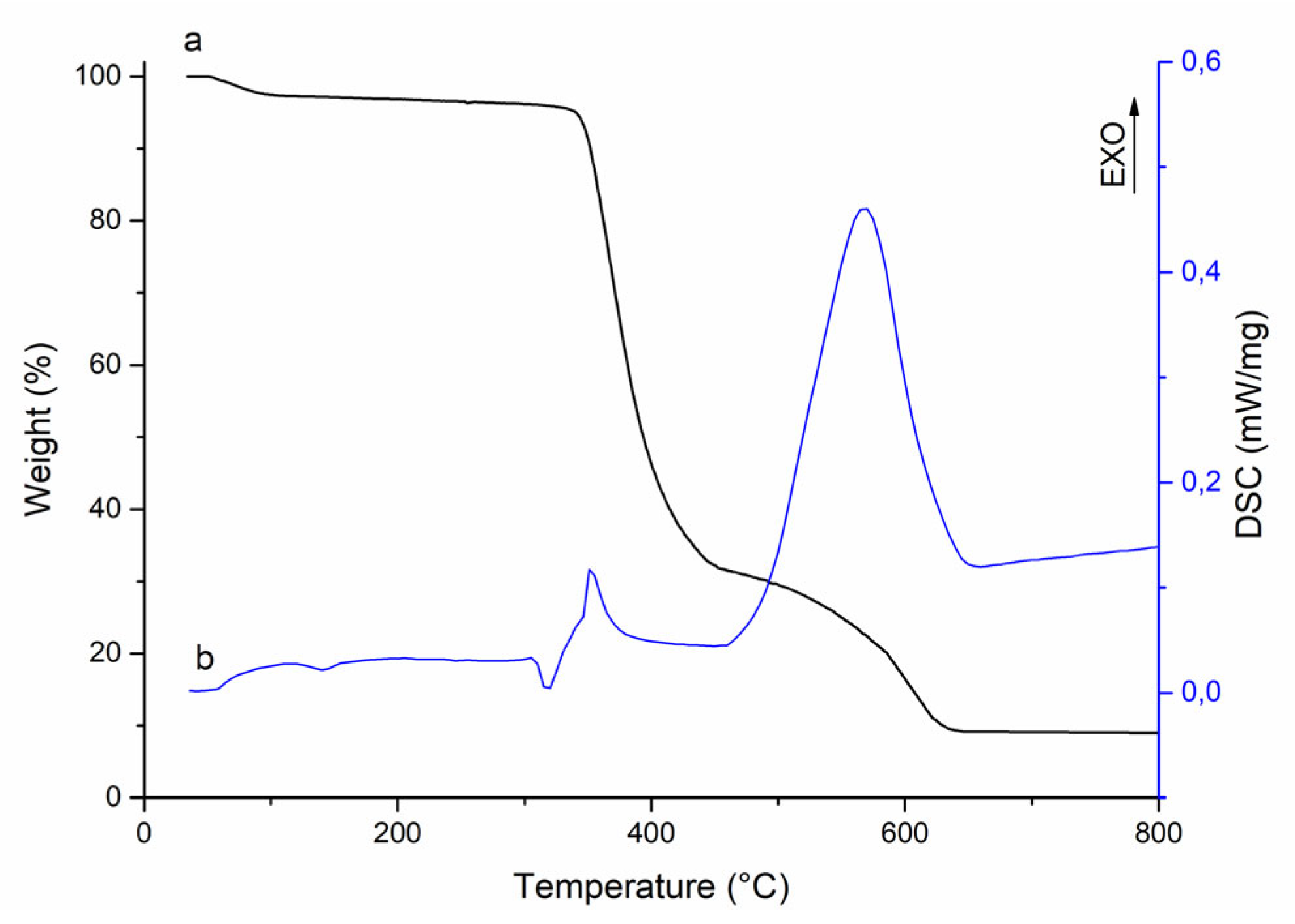
Thermal oxidative degradation of P1–P3 poly(VT-
co-VK) copolymers was studied by thermogravimetric analysis and differential scanning calorimetry. At 50–115 °C, the adsorbed water was released, accompanied by an endothermic effect and a weight loss of 3% (
Figure 3).
In the range of 330–450 °C, thermal degradation of the copolymer was observed, which was accompanied by an exothermic effect with a maximum at 335 °C. The weight loss of the sample in this case was 64%. The triazole and carbazole rings were abstracted with subsequent oxidation to H2O, NO, and CO2. The next stage of polymer degradation was at 470–640 °C. The weight loss of the copolymer was 22%. This stage was characterized by the burning out of the carbon skeleton of the polymer chain of the copolymer and the release of C, NO, and CO2 as a result of the oxidation of decomposition products. The study of thermal oxidative degradation indicated that the thermal stability of the copolymers ranged from 330 to 350 °C.
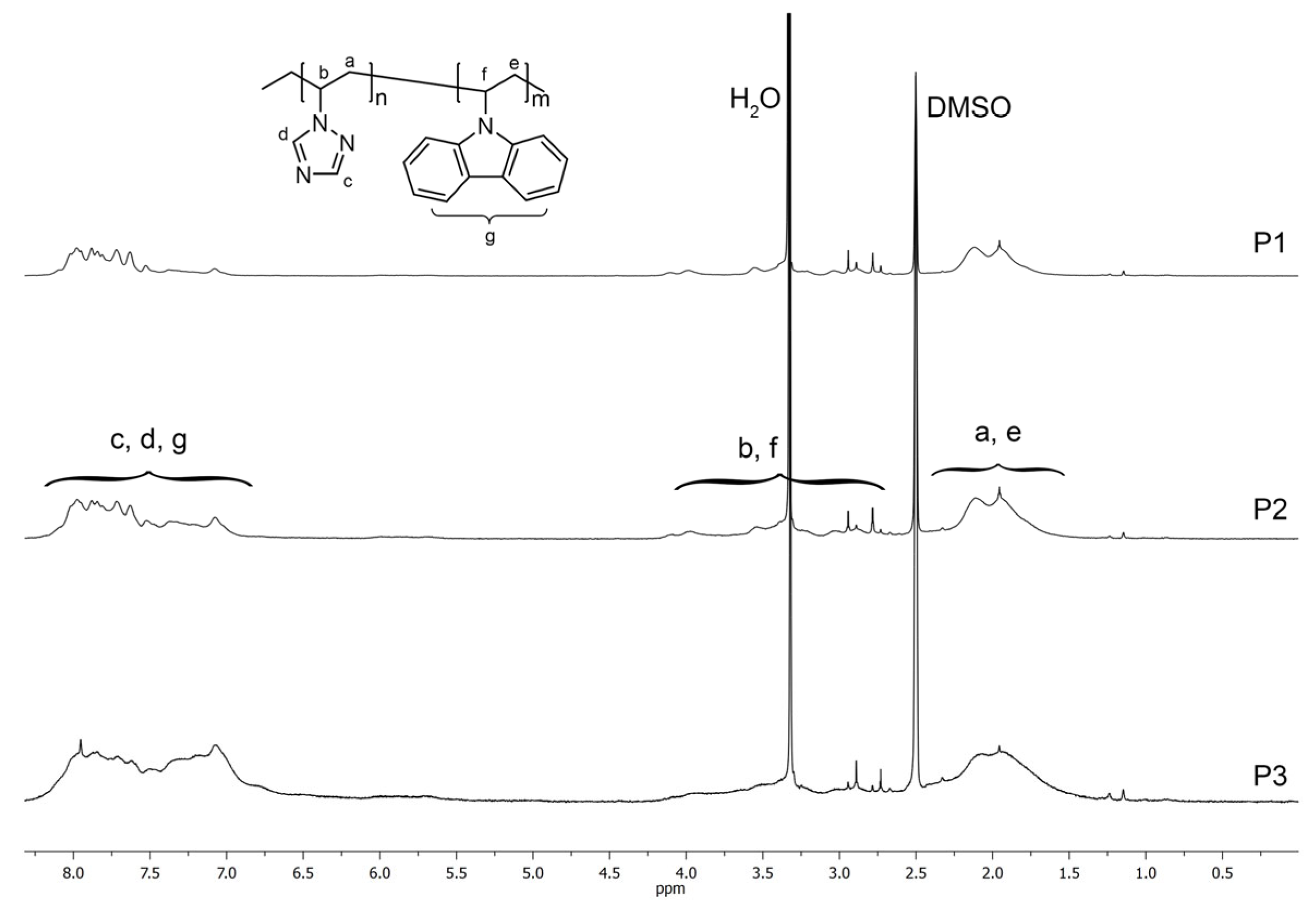
In the
1H NMR spectra of the obtained P1–P3 copolymers, there were no signals of the protons of the
N-vinyl group, but broadened signals of the protons of the methylene groups of the main polymer chain were observed in the region of 1.40–2.25 ppm (a, e), and broad signals of methine protons of the main polymer chain at 2.6–4.15 ppm (b, f) (
Figure 4). Broadened signals of triazole ring protons were observed in the region of 7.50–8.15 ppm (c, d), while strongly broadened signals of carbazole ring protons resonated in the region of 7.50–8.15 ppm (g).
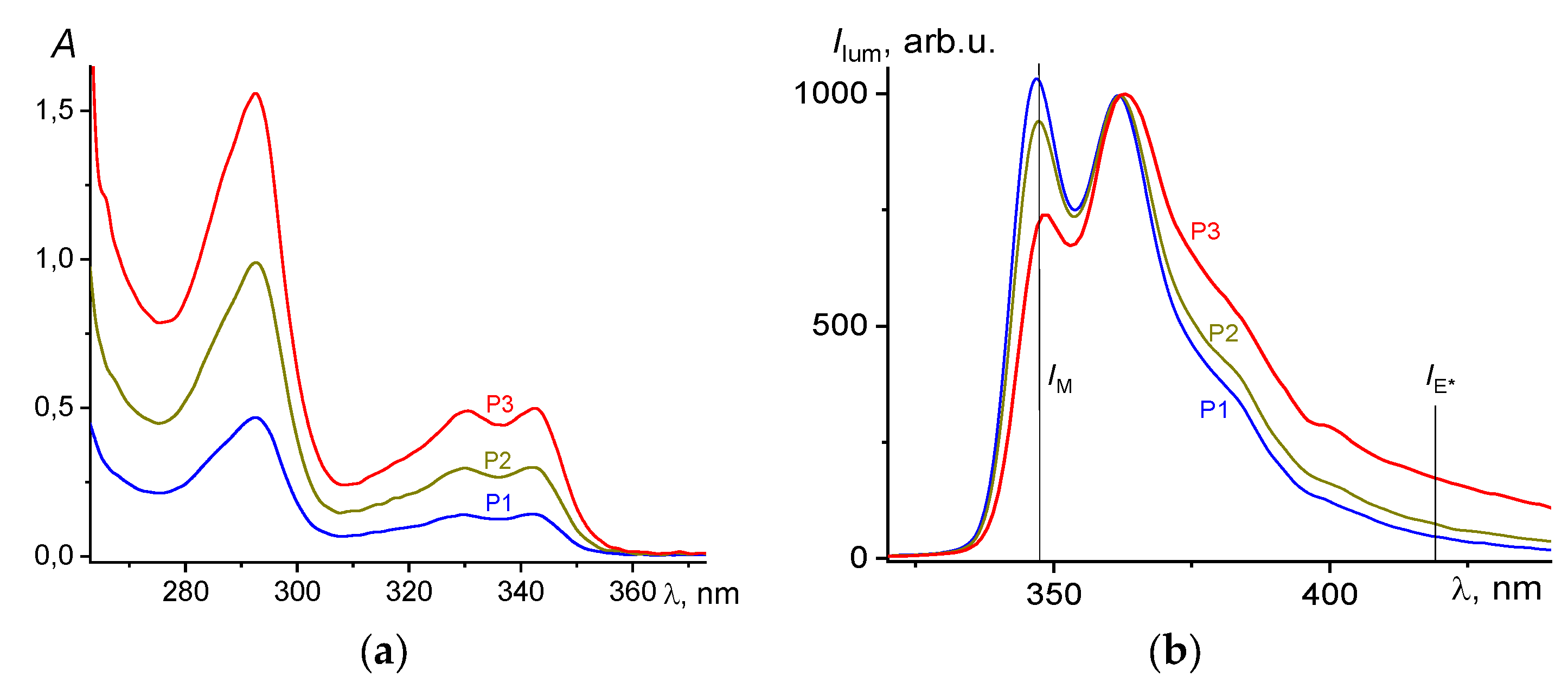
Figure 5 shows the absorption and luminescence spectra of P1–P3 poly(VT-
co-VK) copolymers, depending on the content of VK units in DMF. The content of VK units in the copolymer was in good agreement with the absorption of these units in the region of 320–360 nm (
Figure 5a). In the luminescence spectra in solution, individual VK monomer units appeared at about 350 nm. It is known [
33] that the luminescence spectrum of polyvinylcarbazole (PVK) is mainly represented by excimer luminescence, the short-wavelength band (365–370 nm) that belongs to excimers formed due to the interaction of partially overlapping VK groups, and the long-wavelength (400–420 nm) band belongs to excimers formed by the interaction of completely overlapping VK groups. The band with a maximum at 350 nm, corresponding to the luminescence of individual VK units, practically disappeared for the homopolymer.
In the case of normalization of the emission spectra at 360 nm in the luminescence region of high-energy excimers, it is clearly seen (
Figure 5b) that already in solution, with an increase in the local concentration of VK units due to an increase in their molar content in the polymer chain, the contribution of monomer units decreased and the contribution of luminescence increased low-energy excimers in the region of 400–420 nm.
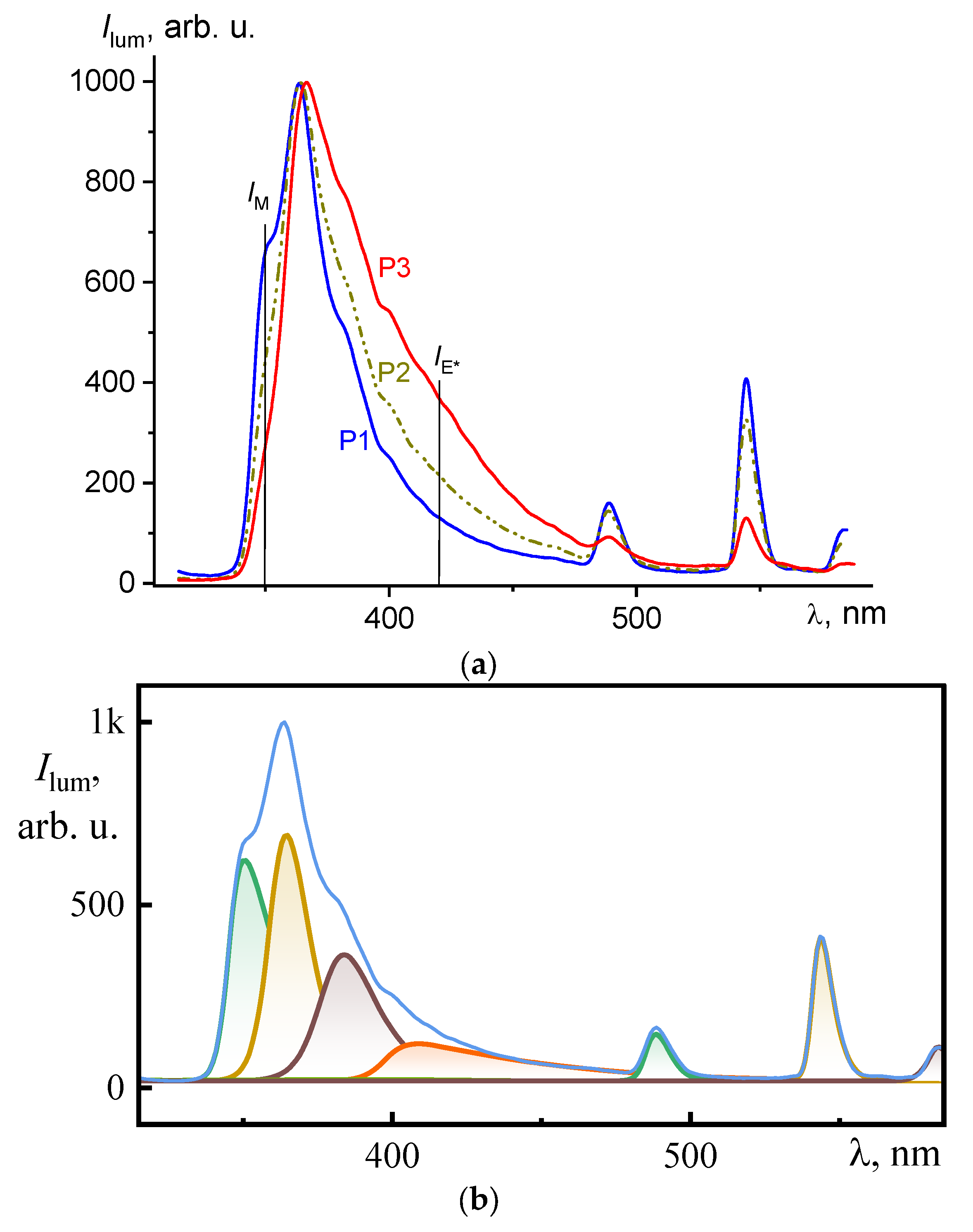
Figure 6a shows the photoluminescence spectra of the obtained MPCs based on Tb
3+ ions and synthesized 1-vinyl-1,2,4-triazole copolymers with a variable content of
N-vinylcarbazole units with hole conductivity (9, 16, and 37 mol.%) in the matrix PMMA. In fluorescent mode (the emission measurement time window starts from zero), for all poly(VT-
co-VK) compositions, two luminescence regions were observed: a short-wavelength one with a wideband from 340 to 450 nm and a long-wavelength one consisting of three distinct bands with a maxima at 489, 544, and 587 nm.
An analysis of the photoluminescence spectra strongly suggests that the emission bands present in the long-wavelength region could be associated with the corresponding
5D
4—
7F
6,
5D
4—
7F
5, и
5D
4—
7F
4 transitions of the electrons of the Tb
3+ ion (in the notation of the Russell−Saunders terms) [
14,
15]. At the same time, photoluminescence of the vinylcarbazole component was observed in the short-wavelength region [
33].
For individual Tb
3+ ions dispersed in a PMMA matrix at the appropriate concentration, their luminescence signal was not observed under similar measurement conditions, thus it can be said that the polymeric ligand poly(VT-
co-VK) sensitized the luminescence of the lanthanide ion, playing the role of a molecular antenna. Moreover, in this case, the energy of electronic excitation was transferred from the units of the VK that absorbed exciting light to Tb
3+ ions by a induction-resonance dipole−dipole mechanism—the Förster resonant energy transfer (FRET)—up to 6 nm [
14].
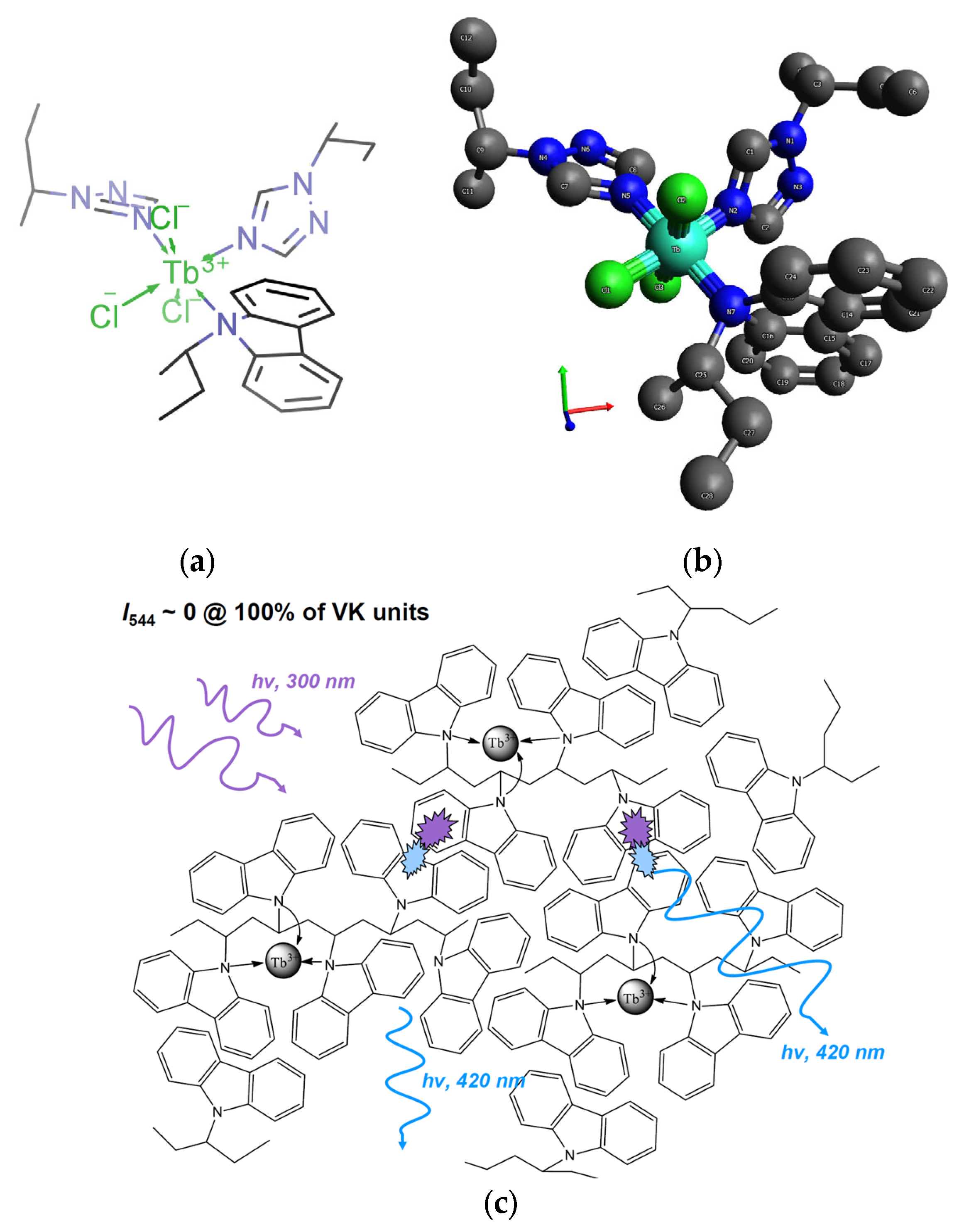
Figure 7a,b shows the possible model explaining the metal−polymeric complex formation locally.
It is known that excimers are “traps” [
35] for exciting light, and FRET is ineffective in such systems. Therefore, in PMMA films containing the homopolymer PVK and Tb
3+, the Tb
3+ luminescence intensity is ~5 rel. units [
36].
Figure 7c explains in general terms the probable channel of energy dissipation through the excimer emission of VK units, which does not allow for the emission of Tb
3+ ions in the {Tb
3+/PVK} system.
The luminescence spectra of metal−polymer complexes in PMMA film were deconvolved to consider the contributions of luminescence bands of different luminescent systems: individual VK units, their excimers, and Tb
3+ in MPC. By deconvolving the emission spectra (
Figure 6), an asymmetric double sigmoidal
Asym2Sig function [
34] was used to determine the full width contribution at half-maximum (FWHM):
where
y₀ is the displacement,
xc is the center of gravity,
A is the amplitude,
w₁ is the full width of the half maximum,
w₂ is the dispersion of the low-energy side, and
w₃ is the dispersion of the high-energy side. The lower bounds were for
w₁ > 0.0,
w₂ > 0.0, and
w₃ > 0.0 when approximated.
Each spectrum was processed using the sum of
Asym2Sig:
where
n is the number of approximated peaks in the absorption band, and
B is the total background for all
Asym2Sigk, standing for the sum of
. The obtained values are given in
Figure S1 (see Electronic Support Materials’ file—ESM). In
Figure 6b, we present the deconvolution for the film in which poly(VT-
co-VK) with 9 mol. % of VK units was used (
Figure S1 in ESM).
Taking into account the above, it is possible to estimate the contribution, %
IE*, in the photoluminescence spectrum for the MPC in the PMMA matrix (
Figure 6) using the cumulative area under the curves of the peaks corresponding to high- and low-energy excimers [VK∙∙∙VK]* with centers of gravity,
xc, at ca. 364, 388, 420, and 460 nm (
Figure 6;
Figure S1 and
Table S1, see in ESM). The photoluminescence peak for VK units had the value of
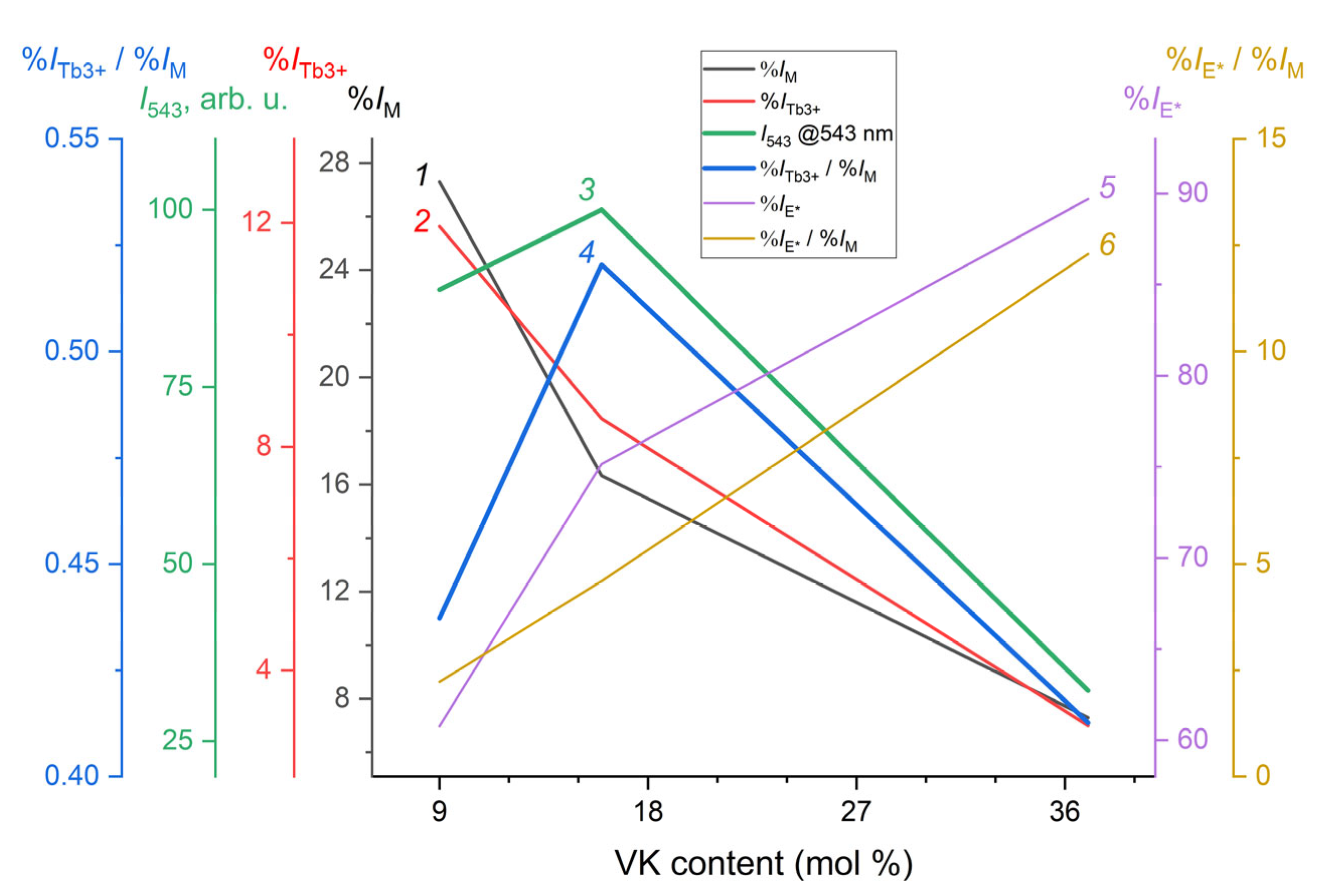
xc in the region of 352–357 nm. The results indicate that augmenting the excimer luminescence contribution, calculated as the ratio of the %
IE* value to the value of the VK unit luminescence contribution, %
IM, intensified with the rise in VK unit content within the copolymer (
Figure 8, curves 5 and 6). The value of %
IE*/%
IM characterized the electronic excitation energy transfer (EET) rate in photoprocesses in PMMA films containing the MPC based on Tb
3+ and poly(VT-
co-VK) (
Table S1 in ESM). The increase in the latter value arose due to the effective overlap of the VK–VK units in an excited state.
The Tb
3+ luminescence contribution, %
ITb(III), decreased symbatically with the decreasing %
IM value, which allowed us to conclude that the electronic excitation energy was transferred from the excited structural units of VK to the lanthanide ion (
Figure 8, curves 1 and 2). However, it was found that as the content of VK units increased in the copolymer, the luminescence intensity
I544 in the
5D
4—
7F
5 transition peak for Tb
3+ passed through a maximum at approximately 16 mol. % of VK units (
Figure 8, curve
3). A further increase in the content of VK units led to a significant decrease in the luminescence intensity of
I544, while the efficiency of the excimer formation according to the ratio of %
IE*/%
IM increased by approximately 2.7 times (
Figure 8, cf. curve 3 and 6;
Table S1 in ESM). The results presented above are related to the sensitizing effect of VK units on Tb
3+ luminescence, i.e., to the so-called molecular antenna effect. The point is that the efficiency of antenna effect for Tb
3+ decreased with an increase in the probability of excimer formation characterized by %
IE*/%
IM: the energy of electronic excitation of VK units was not spent on the photoluminescence of Tb
3+ ions, but fell into excimer traps. It should be noted that the value of %
ITb(III)/%
IM symbatically changed with the value of
I544. The latter fact allowed us to attribute this ratio as a value characterizing the enhancement of the electronic excitation energy transfer from monomeric VK units to lanthanide ions—Tb
3+ luminescence EET rate (
Figure 8, cf. curve 3 and 4). As a result, it appears that the energy transfer to Tb
3+ was most effective at 16 mol. % of the VK units.
It is interesting to note that the appearance of the maximum in the curve for
I544 and its absence in the curve for %
ITb(III) was related to the fact that the antenna effect first increased with increasing the VK content and the constant Tb
3+ content (
Figure 8, curve 2 and 3) at a low content of VK units in the copolymer. At this stage, the excimer formation efficiency was low: the %
IE*/%
IM value was only 2.2, while at its maximum it was twice as high (
Table S1 in ESM). In other words, at this stage, the VK units were more likely to find lanthanide ions and transfer electron excitation energy to them than to find a similar unit removed along the chain or due to an intermacromolecular interaction to form an excimer [VK∙∙∙VK]* in the excited state. However, the contribution of %
ITb(III) decreased steadily depending on the content of VK units, which suggests, together with the above, that there were Tb(III) ions both bound and unbound to VK units in the system. When near each Tb
3+ (ca. 6 nm) there was a VK unit, and “free” Tb
3+ practically did not remain in the system, we observed a transition to a decrease of
I544 value. The value of %
IE*/%
IM, which reflects the efficiency of the electron excitation energy transfer from VK units to Tb
3+, just reached its maximum at this content of VK units. With a further increase in the content of VK units in the system, all the excitation energy was spent on excimer formation.
This result was in good agreement with previously obtained data [
36] on a similar effect of 12–21 mol. % of VK units in copolymers of
N-vinylpyrrolidone and
N-vinylcaprolactam, which manifested itself in an increase in the luminescence intensity of terbium ions by almost two orders of magnitude compared with its luminescence intensity in the presence of copolymers with a higher content of
N-vinylcarbazole units. This effect was due to the competition of two photophysical processes (the formation of excimers and the transfer of electronic excitation energy) proceeding in opposite directions.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}