
Role of Kir4.1 Channel in Auditory Function: Impact on Endocochlear Potential and Hearing Loss
 , ,
, ,  , , , and
, , , and
Abstract
:Featured Application
Abstract
1. Introduction
2. Materials and Methods
3. Results
Data Collection and Screening
4. Discussion
4.1. Auditory Function
Overview of Potassium Channels
- Kir channels are responsible for maintaining the cochlear EP [14];
- Are voltage-gated channels Kv 7.1 (KCNQ1) and Kv 7.4 (KCNQ4), whose mutations have been associated with hearing loss, such as DFNA2 [15]. The resting potential and membrane potential in internal hair cells are regulated by these channels. When opened, they allow for the release of K+ from the cell, which supports the maintenance of the membrane potential of the internal ciliate cell, which is fundamental for hearing transduction [20];
- Large-conductance calcium-activated potassium channels, named BK channels, are capable of allowing significant K+ efflux due to the binding of Ca2+ ions to specific sites in the channel protein, which can hyperpolarize the cell membrane and regulate cellular excitability. BK channels play a role in the transduction of the auditory signal in hair cells by responding to changes in sound intensity [21].
4.2. Kir Channel Family
4.3. Kir4.1 Channel
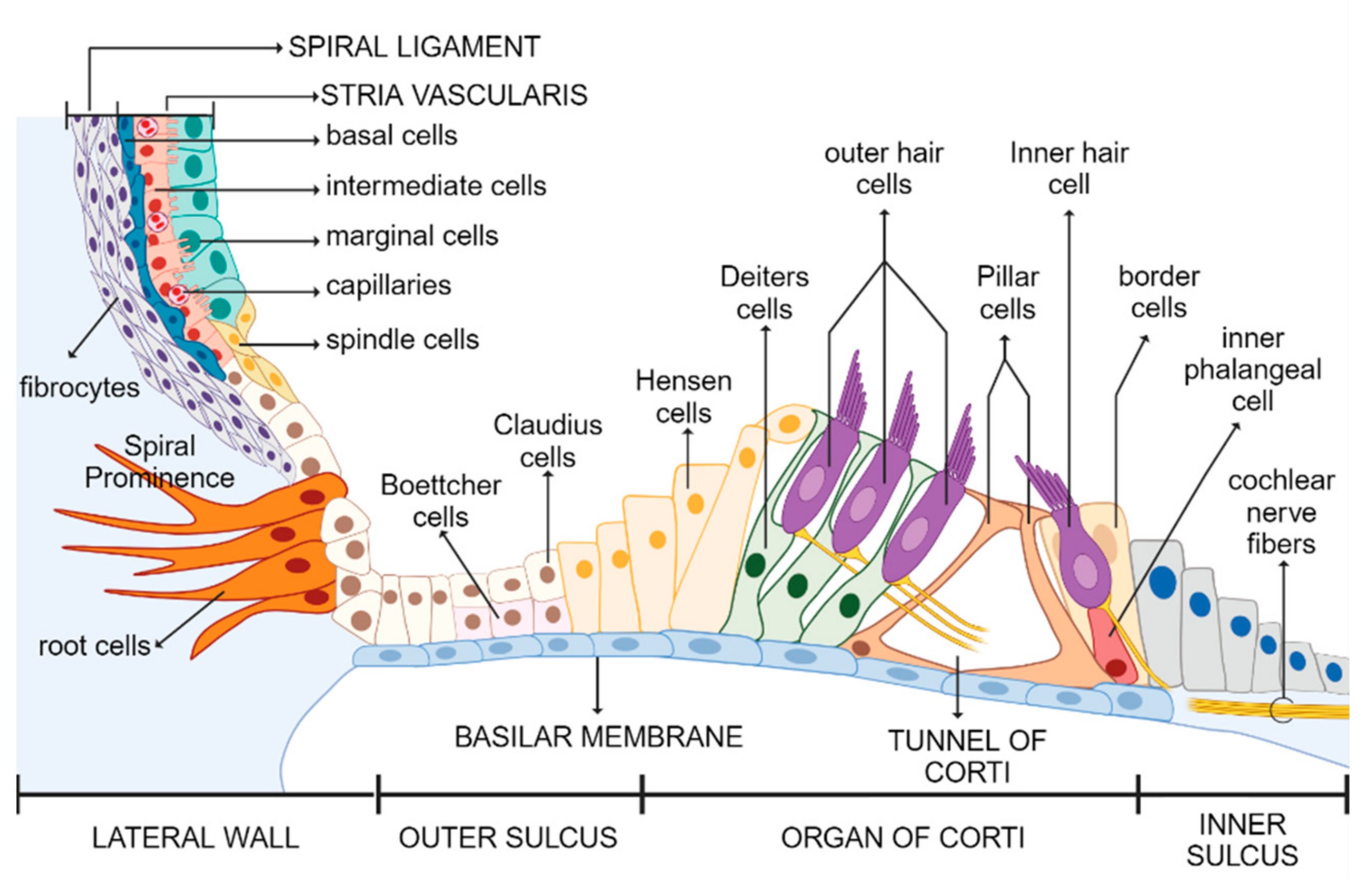
4.3.1. Kir4.1 Channel Expression
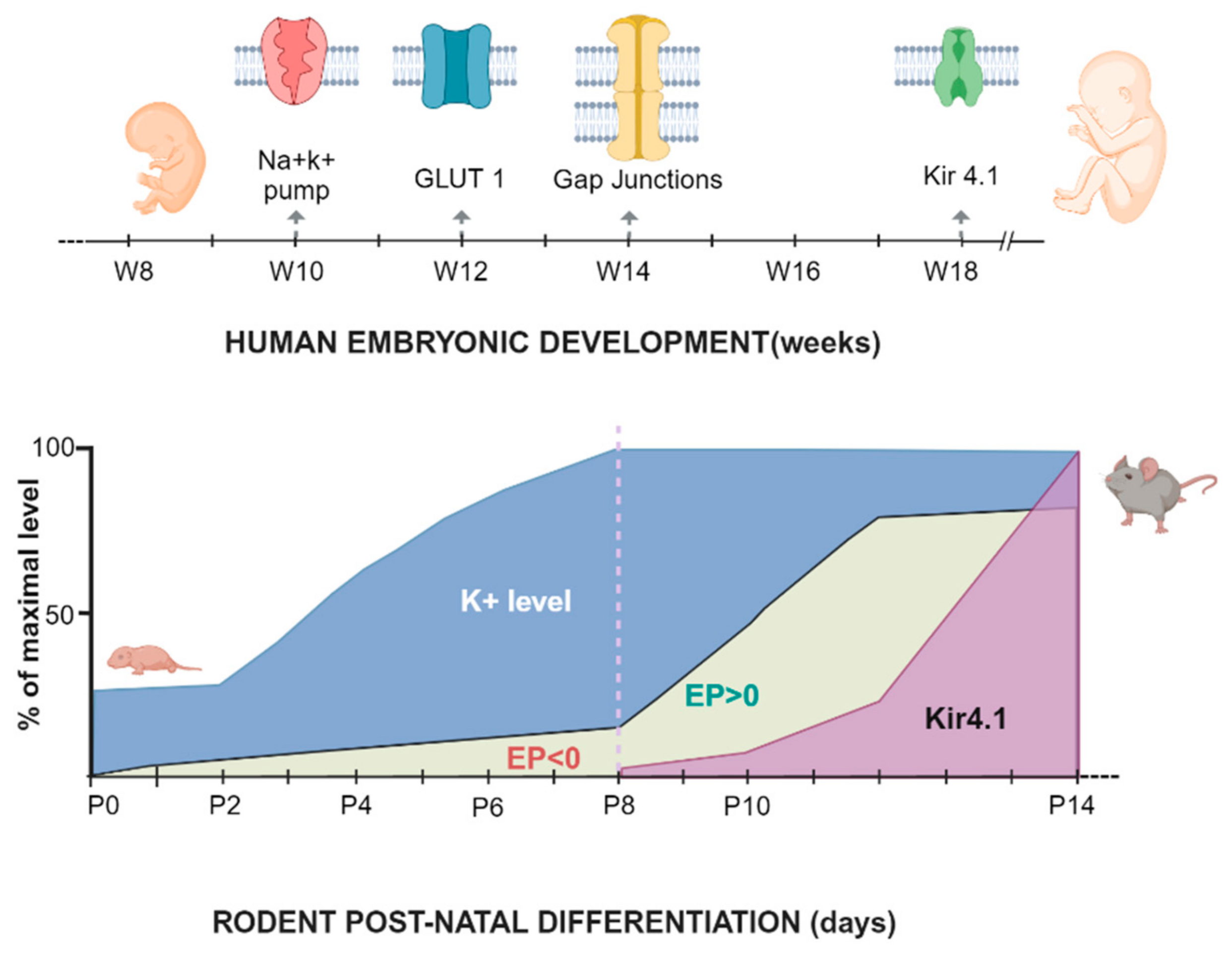
4.3.2. Embryonal and Postnatal Development
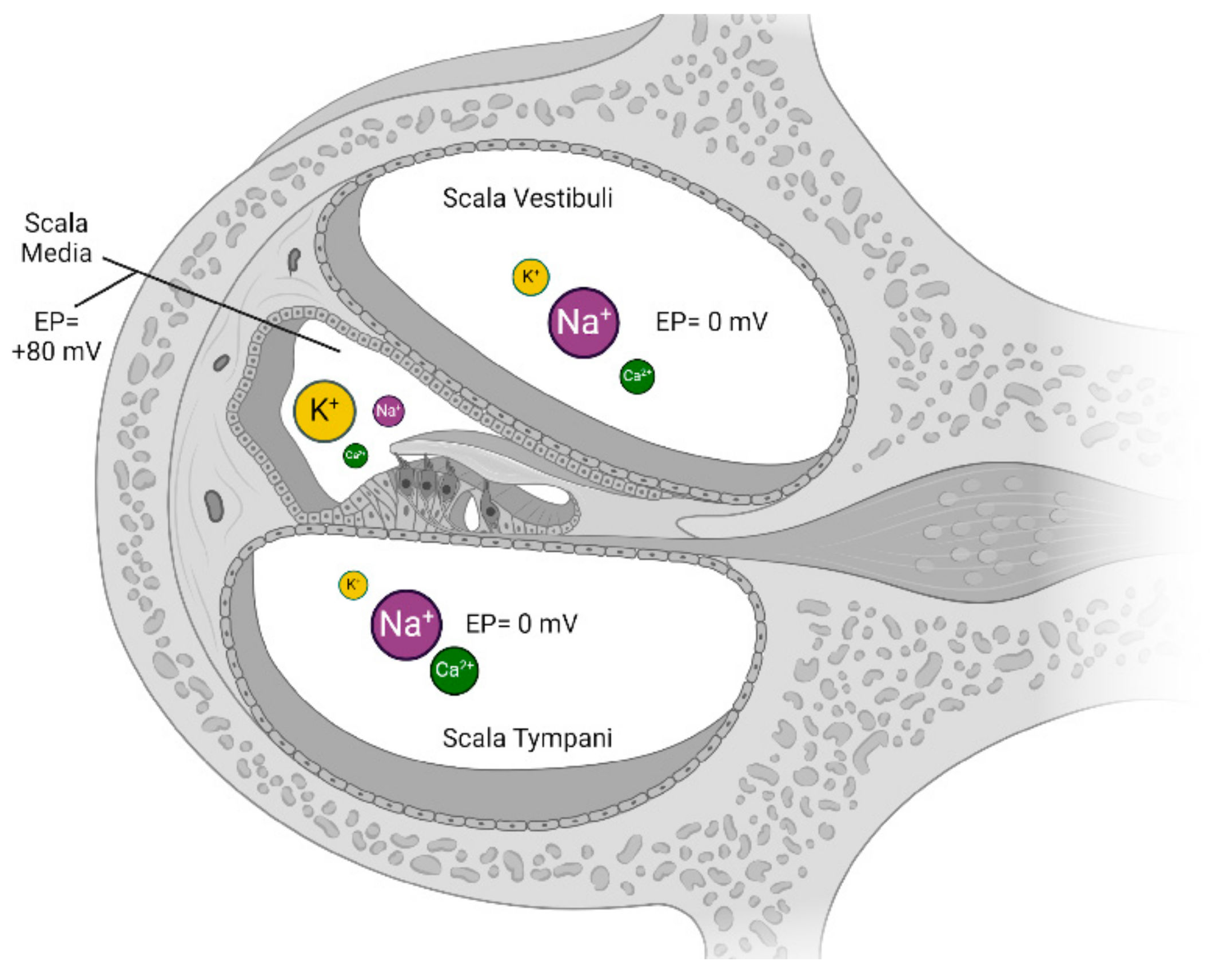
4.3.3. Endocochlear Potential
- Marginal cells actively pump K+ ions into their cytoplasm to maintain a high intracellular K+ concentration;
- This generates a high gradient of K+ between the marginal and intermediate cells, with the intermediate cells having lower intracellular K+;
- Furthermore, the intermediate cells have a relatively negative membrane potential compared to the marginal cells;
- This double electrochemical gradient (chemical for K+ and electrical for the membrane potential) causes K+ to spontaneously efflux through the Kir4.1 channel into the intrastrial space.
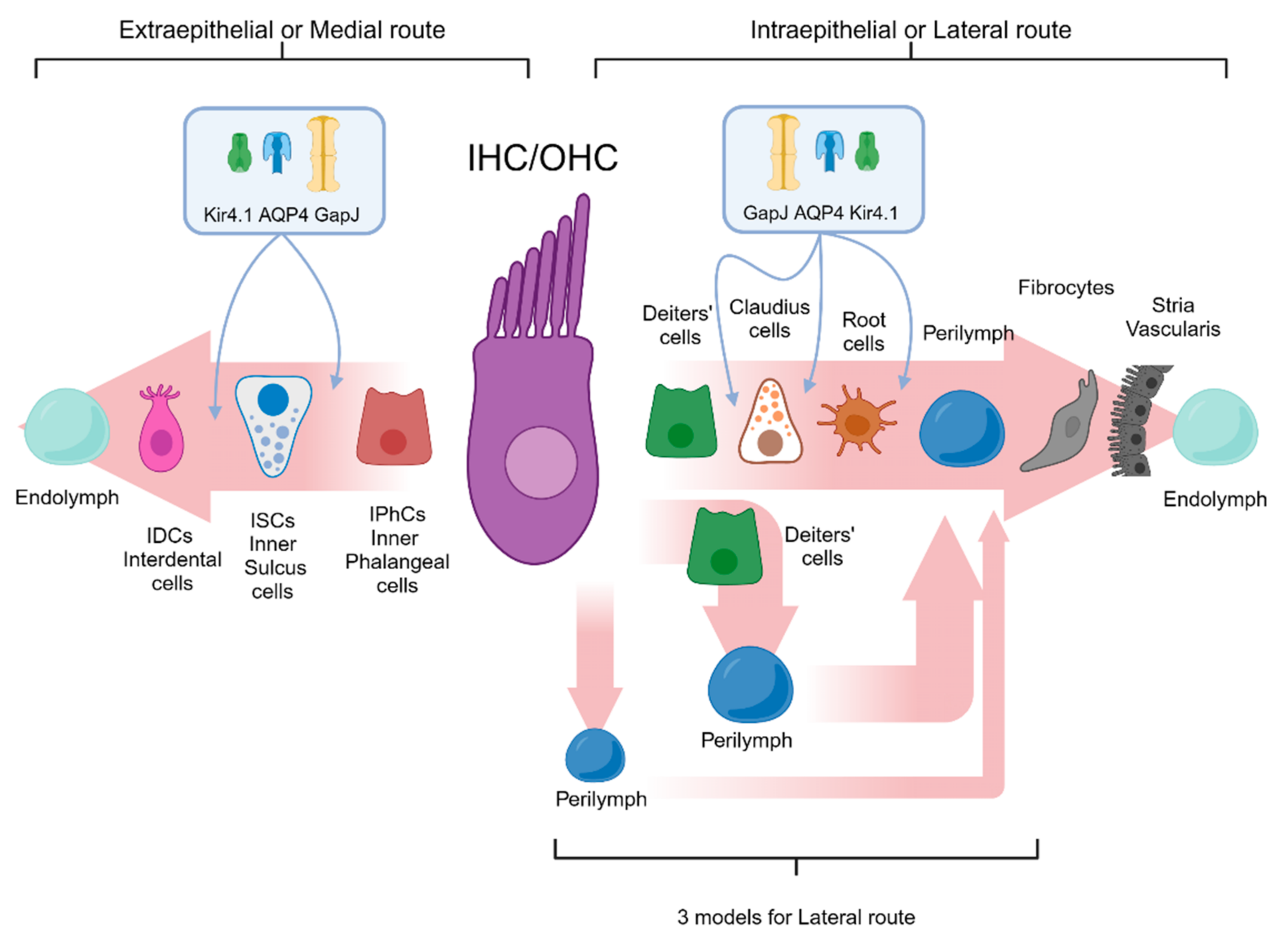
4.3.4. K+ Cochlear Cycle
- Potassium recycling through open perilymph (first pathway): The potassium efflux from the hair cells is directed into the interstitial space between the inner hair cells and supporting cells, as well as between the outer hair cells and Deiters’ cells. Subsequently, potassium is introduced into the perilymph and captured by the fibrocytes of the spiral ligament without the involvement of other cells in its entry into the perilymph.
- Potassium recycling buffered (second pathway): The potassium introduced into the interstitial space, released from the OHCs or IHCs, is promptly recaptured by Deiters’ cells or supporting cells, which mediate its release into the perilymph.
- Potassium recycling via gap junctions (third pathway): Once the IHCs or OHCs release potassium ions into the interstitial space, it is immediately recaptured by Deiters’ cells and supporting cells and subsequently transported back to the stria vascularis via a dense network of gap junctions.
4.3.5. Pathophysiology
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. PubMed
Appendix A.2. Scopus
Appendix A.3. Web of Science
References
- Deafness and Hearing Loss. Available online: https://www.who.int/news-room/fact-sheets/detail/deafness-and-hearing-loss (accessed on 17 April 2024).
- Busi, M.; Castiglione, A.; Taddei Masieri, M.; Ravani, A.; Guaran, V.; Astolfi, L.; Trevisi, P.; Ferlini, A.; Martini, A. Novel Mutations in the SLC26A4 Gene. Int. J. Pediatr. Otorhinolaryngol. 2012, 76, 1249–1254. [Google Scholar] [CrossRef] [PubMed]
- Guaran, V.; Astolfi, L.; Castiglione, A.; Simoni, E.; Olivetto, E.; Galasso, M.; Trevisi, P.; Busi, M.; Volinia, S.; Martini, A. Association between Idiopathic Hearing Loss and Mitochondrial DNA Mutations: A Study on 169 Hearing-Impaired Subjects. Int. J. Mol. Med. 2013, 32, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Bernardinelli, E.; Roesch, S.; Simoni, E.; Marino, A.; Rasp, G.; Astolfi, L.; Sarikas, A.; Dossena, S. Novel POU3F4 Variants Identified in Patients with Inner Ear Malformations Exhibit Aberrant Cellular Distribution and Lack of SLC6A20 Transcriptional Upregulation. Front. Mol. Neurosci. 2022, 15, 999833. [Google Scholar] [CrossRef] [PubMed]
- Korver, A.M.H.; Smith, R.J.H.; Van Camp, G.; Schleiss, M.R.; Bitner-Glindzicz, M.A.K.; Lustig, L.R.; Usami, S.-I.; Boudewyns, A.N. Congenital Hearing Loss. Nat. Rev. Dis. Primer 2017, 3, 16094. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, H.; Okubo, T.; Shinagawa, J.; Nishio, S.-Y.; Takumi, Y.; Usami, S.-I. Epidemiology, Aetiology and Diagnosis of Congenital Hearing Loss via Hearing Screening of 153913 Newborns. Int. J. Epidemiol. 2024, 53, dyae052. [Google Scholar] [CrossRef]
- Locher, H.; de Groot, J.C.M.J.; van Iperen, L.; Huisman, M.A.; Frijns, J.H.M.; Chuva de Sousa Lopes, S.M. Development of the Stria Vascularis and Potassium Regulation in the Human Fetal Cochlea: Insights into Hereditary Sensorineural Hearing Loss. Dev. Neurobiol. 2015, 75, 1219–1240. [Google Scholar] [CrossRef] [PubMed]
- Franz, L.; Incognito, A.; Gallo, C.; Turolla, L.; Scquizzato, E.; Cenedese, R.; Matarazzo, A.; Savegnago, D.; Zanatta, P.; Genovese, E.; et al. Audiological Phenotypes of Connexin Gene Mutation Patterns: A Glance at Different GJB2/GJB6 Gene Mutation Profiles. Children 2024, 11, 194. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Vallon, V.; Knipper, M.; Wangemann, P. Functional Significance of Channels and Transporters Expressed in the Inner Ear and Kidney. Am. J. Physiol.-Cell Physiol. 2007, 293, C1187–C1208. [Google Scholar] [CrossRef] [PubMed]
- Jagger, D.J.; Forge, A. Connexins and Gap Junctions in the Inner Ear—It’s Not Just about K+ Recycling. Cell Tissue Res. 2015, 360, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Hibino, H.; Nin, F.; Tsuzuki, C.; Kurachi, Y. How Is the Highly Positive Endocochlear Potential Formed? The Specific Architecture of the Stria Vascularis and the Roles of the Ion-Transport Apparatus. Pflug. Arch.-Eur. J. Physiol. 2010, 459, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Casale, J.; Kandle, P.F.; Murray, I.V.; Murr, N.I. Physiology, Cochlear Function. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Rask-Andersen, H.; Liu, W.; Erixon, E.; Kinnefors, A.; Pfaller, K.; Schrott-Fischer, A.; Glueckert, R. Human Cochlea: Anatomical Characteristics and Their Relevance for Cochlear Implantation. Anat. Rec. 2012, 295, 1791–1811. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhao, H.-B. The Role of an Inwardly Rectifying K+ Channel (Kir4.1) in the Inner Ear and Hearing Loss. Neuroscience 2014, 265, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Zdebik, A.A.; Wangemann, P.; Jentsch, T.J. Potassium Ion Movement in the Inner Ear: Insights from Genetic Disease and Mouse Models. Physiology 2009, 24, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Takiguchi, Y.; Sun, G.; Ogawa, K.; Matsunaga, T. Long-Lasting Changes in the Cochlear K+ Recycling Structures after Acute Energy Failure. Neurosci. Res. 2013, 77, 33–41. [Google Scholar] [CrossRef] [PubMed]
- MacKinnon, R. Potassium Channels. FEBS Lett. 2003, 555, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, S.D.; Kidd, J.F.; Law, R.J.; Franks, C.J.; Sattelle, D.B. Structure and Function of Two-Pore-Domain K+ Channels: Contributions from Genetic Model Organisms. Trends Pharmacol. Sci. 2005, 26, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Q.; Purhonen, P.; Hebert, H. Structure of Potassium Channels. Cell. Mol. Life Sci. 2015, 72, 3677–3693. [Google Scholar] [CrossRef] [PubMed]
- Robbins, J. KCNQ Potassium Channels: Physiology, Pathophysiology, and Pharmacology. Pharmacol. Ther. 2001, 90, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Pyott, S.J.; Duncan, R.K. Chapter Ten—BK Channels in the Vertebrate Inner Ear. In International Review of Neurobiology; Contet, C., Ed.; Big on Bk; Academic Press: Cambridge, MA, USA, 2016; Volume 128, pp. 369–399. [Google Scholar]
- Bazard, P.; Frisina, R.D.; Acosta, A.A.; Dasgupta, S.; Bauer, M.A.; Zhu, X.; Ding, B. Roles of Key Ion Channels and Transport Proteins in Age-Related Hearing Loss. Int. J. Mol. Sci. 2021, 22, 6158. [Google Scholar] [CrossRef] [PubMed]
- Fakler, B.; Ruppersberg, J.P. Functional and Molecular Diversity Classifies the Family of Inward-Rectifier K+ Channels. Cell Physiol. Biochem. 1996, 6, 195–209. [Google Scholar] [CrossRef]
- Parrock, S.; Hussain, S.; Issler, N.; Differ, A.-M.; Lench, N.; Guarino, S.; Oosterveld, M.J.S.; Keijzer-Veen, M.; Brilstra, E.; van Wieringen, H.; et al. KCNJ10 Mutations Display Differential Sensitivity to Heteromerisation with KCNJ16. Nephron Physiol. 2013, 123, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.M.; Lopes, C.M.B.; Rosenhouse-Dantsker, A.; Connelly, H.L.; Matavel, A.; O-Uchi, J.; McBeath, E.; Gray, D.A. Molecular Basis of Decreased Kir4.1 Function in SeSAME/EAST Syndrome. J. Am. Soc. Nephrol. JASN 2010, 21, 2117–2129. [Google Scholar] [CrossRef] [PubMed]
- Sodium-Potassium-Chloride Cotransport. Available online: https://journals.physiology.org/doi/epdf/10.1152/physrev.2000.80.1.211 (accessed on 29 April 2024).
- Strepay, D.; Olszewski, R.T.; Nixon, S.; Korrapati, S.; Adadey, S.; Griffith, A.J.; Su, Y.; Liu, J.; Vishwasrao, H.; Gu, S.; et al. Transgenic Tg(Kcnj10-ZsGreen) Fluorescent Reporter Mice Allow Visualization of Intermediate Cells in the Stria Vascularis. Sci. Rep. 2024, 14, 3038. [Google Scholar] [CrossRef]
- Hibino, H.; Horio, Y.; Inanobe, A.; Doi, K.; Ito, M.; Yamada, M.; Gotow, T.; Uchiyama, Y.; Kawamura, M.; Kubo, T.; et al. An ATP-Dependent Inwardly Rectifying Potassium Channel, KAB-2 (Kir4. 1), in Cochlear Stria Vascularis of Inner Ear: Its Specific Subcellular Localization and Correlation with the Formation of Endocochlear Potential. J. Neurosci. Off. J. Soc. Neurosci. 1997, 17, 4711–4721. [Google Scholar] [CrossRef] [PubMed]
- Wangemann, P.; Itza, E.M.; Albrecht, B.; Wu, T.; Jabba, S.V.; Maganti, R.J.; Ho Lee, J.; Everett, L.A.; Wall, S.M.; Royaux, I.E.; et al. Loss of KCNJ10 Protein Expression Abolishes Endocochlear Potential and Causes Deafness in Pendred Syndrome Mouse Model. BMC Med. 2004, 2, 30. [Google Scholar] [CrossRef] [PubMed]
- Korrapati, S.; Taukulis, I.; Olszewski, R.; Pyle, M.; Gu, S.; Singh, R.; Griffiths, C.; Martin, D.; Boger, E.; Morell, R.J.; et al. Single Cell and Single Nucleus RNA-Seq Reveal Cellular Heterogeneity and Homeostatic Regulatory Networks in Adult Mouse Stria Vascularis. Front. Mol. Neurosci. 2019, 12, 316. [Google Scholar] [CrossRef] [PubMed]
- Jagger, D.J.; Nevill, G.; Forge, A. The Membrane Properties of Cochlear Root Cells Are Consistent with Roles in Potassium Recirculation and Spatial Buffering. JARO—J. Assoc. Res. Otolaryngol. 2010, 11, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Olszewski, R.; Taukulis, I.; Wei, Z.; Martin, D.; Morell, R.J.; Hoa, M. Characterization of Rare Spindle and Root Cell Transcriptional Profiles in the Stria Vascularis of the Adult Mouse Cochlea. Sci. Rep. 2020, 10, 18100. [Google Scholar] [CrossRef] [PubMed]
- Rozengurt, N.; Lopez, I.; Chiu, C.-S.; Kofuji, P.; Lester, H.A.; Neusch, C. Time Course of Inner Ear Degeneration and Deafness in Mice Lacking the Kir4.1 Potassium Channel Subunit. Hear. Res. 2003, 177, 71–80. [Google Scholar] [CrossRef]
- Tang, X.; Schmidt, T.M.; Perez-Leighton, C.E.; Kofuji, P. Kir4.1 Is Responsible for the Native Inward Potassium Conductance of Satellite Glial Cells in Sensory Ganglia. Neuroscience 2010, 166, 397. [Google Scholar] [CrossRef] [PubMed]
- Lorente-Cánovas, B.; Ingham, N.; Norgett, E.E.; Golder, Z.J.; Karet Frankl, F.E.; Steel, K.P. Mice Deficient in H+-ATPase A4 Subunit Have Severe Hearing Impairment Associated with Enlarged Endolymphatic Compartments within the Inner Ear. Dis. Model. Mech. 2013, 6, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.-Y.; Affortit, C.; Ranum, P.T.; West, C.; Walls, W.D.; Yoshimura, H.; Shao, J.Q.; Mostaert, B.; Smith, R.J.H. Single-Cell RNA-Sequencing of Stria Vascularis Cells in the Adult Slc26a4−/− Mouse. BMC Med. Genom. 2023, 16, 133. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Ulfendahl, M.; Järlebark, L. Spatiotemporal Loss of K+ Transport Proteins in the Developing Cochlear Lateral Wall of Guinea Pigs with Hereditary Deafness. Eur. J. Neurosci. 2008, 27, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Wei, D.; Järlebark, L. Developmental Expression and Localization of KCNJ10 K+ Channels in the Guinea Pig Inner Ear. Neuroreport 2006, 17, 475–479. [Google Scholar] [CrossRef] [PubMed]
- De Iriarte Rodríguez, R.; Magariños, M.; Pfeiffer, V.; Rapp, U.R.; Varela-Nieto, I. C-Raf Deficiency Leads to Hearing Loss and Increased Noise Susceptibility. Cell. Mol. Life Sci. 2015, 72, 3983–3998. [Google Scholar] [CrossRef]
- Kim, H.J.; Gratton, M.A.; Lee, J.-H.; Perez Flores, M.C.; Wang, W.; Doyle, K.J.; Beermann, F.; Crognale, M.A.; Yamoah, E.N. Precise Toxigenic Ablation of Intermediate Cells Abolishes the “Battery” of the Cochlear Duct. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 14601–14606. [Google Scholar] [CrossRef] [PubMed]
- Cazals, Y.; Bévengut, M.; Zanella, S.; Brocard, F.; Barhanin, J.; Gestreau, C. KCNK5 Channels Mostly Expressed in Cochlear Outer Sulcus Cells Are Indispensable for Hearing. Nat. Commun. 2015, 6, 8780. [Google Scholar] [CrossRef] [PubMed]
- Hibino, H.; Higashi-Shingai, K.; Fujita, A.; Iwai, K.; Ishii, M.; Kurachi, Y. Expression of an Inwardly Rectifying K+ Channel, Kir5.1, in Specific Types of Fibrocytes in the Cochlear Lateral Wall Suggests Its Functional Importance in the Establishment of Endocochlear Potential. Eur. J. Neurosci. 2004, 19, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Marcus, D.C.; Wu, T.; Wangemann, P.; Kofuji, P. KCNJ10 (Kir4.1) Potassium Channel Knockout Abolishes Endocochlear Potential. Am. J. Physiol.-Cell Physiol. 2002, 282, C403–C407. [Google Scholar] [CrossRef] [PubMed]
- Ashmore, J. Cochlear Outer Hair Cell Motility. Physiol. Rev. 2008, 88, 173–210. [Google Scholar] [CrossRef] [PubMed]
- Spitzmaul, G.; Rías, E.; Dionisio, L.; Spitzmaul, G.; Rías, E.; Dionisio, L. Potential Mechanisms of Hearing Loss Due to Impaired Potassium Circulation in the Organ of Corti. In Updates on Hearing Loss and its Rehabilitation; IntechOpen: London, UK, 2023; ISBN 978-1-83769-777-9. [Google Scholar]
- Eckhard, A.; Gleiser, C.; Rask-Andersen, H.; Arnold, H.; Liu, W.; Mack, A.; Müller, M.; Löwenheim, H.; Hirt, B. Co-Localisation of K(Ir)4.1 and AQP4 in Rat and Human Cochleae Reveals a Gap in Water Channel Expression at the Transduction Sites of Endocochlear K(+) Recycling Routes. Cell Tissue Res. 2012, 350, 27–43. [Google Scholar] [CrossRef]
- Eckhard, A.; Löwenheim, H. Water Regulation in the Cochlea: Do Molecular Water Channels Facilitate Potassium-Dependent Sound. Transduction? HNO 2014, 62, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Bockenhauer, D.; Feather, S.; Stanescu, H.C.; Bandulik, S.; Zdebik, A.A.; Reichold, M.; Tobin, J.; Lieberer, E.; Sterner, C.; Landoure, G.; et al. Epilepsy, Ataxia, Sensorineural Deafness, Tubulopathy, and KCNJ10 Mutations. N. Engl. J. Med. 2009, 360, 1960–1970. [Google Scholar] [CrossRef] [PubMed]
- Scholl, U.I.; Choi, M.; Liu, T.; Ramaekers, V.T.; Häusler, M.G.; Grimmer, J.; Tobe, S.W.; Farhi, A.; Nelson-Williams, C.; Lifton, R.P. Seizures, Sensorineural Deafness, Ataxia, Mental Retardation, and Electrolyte Imbalance (SeSAME Syndrome) Caused by Mutations in KCNJ10. Proc. Natl. Acad. Sci. USA 2009, 106, 5842–5847. [Google Scholar] [CrossRef] [PubMed]
- Scholl, U.I.; Dave, H.B.; Lu, M.; Farhi, A.; Nelson-Williams, C.; Listman, J.A.; Lifton, R.P. SeSAME/EAST Syndrome--Phenotypic Variability and Delayed Activity of the Distal Convoluted Tubule. Pediatr. Nephrol. Berl. Ger. 2012, 27, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Freudenthal, B.; Kulaveerasingam, D.; Lingappa, L.; Shah, M.A.; Brueton, L.; Wassmer, E.; Ognjanovic, M.; Dorison, N.; Reichold, M.; Bockenhauer, D.; et al. KCNJ10 Mutations Disrupt Function in Patients with EAST Syndrome. Nephron Physiol. 2011, 119, P40–P48. [Google Scholar] [CrossRef]
- Celmina, M.; Micule, I.; Inashkina, I.; Audere, M.; Kuske, S.; Pereca, J.; Stavusis, J.; Pelnena, D.; Strautmanis, J. EAST/SeSAME Syndrome: Review of the Literature and Introduction of Four New Latvian Patients. Clin. Genet. 2019, 95, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Wangemann, P. The Role of Pendrin in the Development of the Murine Inner Ear. Cell. Physiol. Biochem. 2011, 28, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Gurrola, J.G.; Wu, H.; Chiu, S.M.; Wangemann, P.; Snyder, P.M.; Smith, R.J.H. Mutations of KCNJ10 Together with Mutations of SLC26A4 Cause Digenic Nonsyndromic Hearing Loss Associated with Enlarged Vestibular Aqueduct Syndrome. Am. J. Hum. Genet. 2009, 84, 651–657. [Google Scholar] [CrossRef]
- Wangemann, P.; Nakaya, K.; Wu, T.; Maganti, R.J.; Itza, E.M.; Sanneman, J.D.; Harbidge, D.G.; Billings, S.; Marcus, D.C. Loss of Cochlear HCO3- Secretion Causes Deafness via Endolymphatic Acidification and Inhibition of Ca2+ Reabsorption in a Pendred Syndrome Mouse Model. Am. J. Physiol.-Ren. Physiol. 2007, 292, F1345–F1353. [Google Scholar] [CrossRef]
- Singh, R.; Wangemann, P. Free Radical Stress-Mediated Loss of Kcnj10 Protein Expression in Stria Vascularis Contributes to Deafness in Pendred Syndrome Mouse Model. Am. J. Physiol.-Ren. Physiol. 2008, 294, F139–F148. [Google Scholar] [CrossRef] [PubMed]
- Wangemann, P. Mouse Models for Pendrin-Associated Loss of Cochlear and Vestibular Function. Cell. Physiol. Biochem. 2013, 32, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Bałdyga, N.; Oziębło, D.; Gan, N.; Furmanek, M.; Leja, M.L.; Skarżyński, H.; Ołdak, M. The Genetic Background of Hearing Loss in Patients with EVA and Cochlear Malformation. Genes 2023, 14, 335. [Google Scholar] [CrossRef] [PubMed]
- Landa, P.; Differ, A.-M.; Rajput, K.; Jenkins, L.; Bitner-Glindzicz, M. Lack of Significant Association between Mutations of KCNJ10 or FOXI1 and SLC26A4 Mutations in Pendred Syndrome/Enlarged Vestibular Aqueducts. BMC Med. Genet. 2013, 14, 85. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wang, X.; Sun, L.; Jiang, H. Screening of SLC26A4, FOXI1, KCNJ10, and GJB2 in Bilateral Deafness Patients with Inner Ear Malformation. Otolaryngol. Head Neck Surg. 2012, 146, 972–978. [Google Scholar] [CrossRef]
- Jonard, L.; Niasme-Grare, M.; Bonnet, C.; Feldmann, D.; Rouillon, I.; Loundon, N.; Calais, C.; Catros, H.; David, A.; Dollfus, H.; et al. Screening of SLC26A4, FOXI1 and KCNJ10 Genes in Unilateral Hearing Impairment with Ipsilateral Enlarged Vestibular Aqueduct. Int. J. Pediatr. Otorhinolaryngol. 2010, 74, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Suzumoto, Y.; Columbano, V.; Gervasi, L.; Giunta, R.; Mattina, T.; Trimarchi, G.; Capolongo, G.; Simeoni, M.; Perna, A.F.; Zacchia, M.; et al. A Case Series of Adult Patients Affected by EAST/SeSAME Syndrome Suggests More Severe Disease in Subjects Bearing KCNJ10 Truncating Mutations. Intractable Rare Dis. Res. 2021, 10, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Sala-Rabanal, M.; Kucheryavykh, L.Y.; Skatchkov, S.N.; Eaton, M.J.; Nichols, C.G. Molecular Mechanisms of EAST/SeSAME Syndrome Mutations in Kir4.1 (KCNJ10). J. Biol. Chem. 2010, 285, 36040–36048. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Li, G.; Noble, K.V.; Li, Y.; Barth, J.L.; Schulte, B.A.; Lang, H. Age-Dependent Alterations of Kir4.1 Expression in Neural Crest-Derived Cells of the Mouse and Human Cochlea. Neurobiol. Aging 2019, 80, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Morán-Zendejas, R.; Delgado-Ramírez, M.; Xu, J.; Valdés-Abadía, B.; Aréchiga-Figueroa, I.A.; Cui, M.; Rodríguez-Menchaca, A.A. In Vitro and in Silico Characterization of the Inhibition of Kir4.1 Channels by Aminoglycoside Antibiotics. Br. J. Pharmacol. 2020, 177, 4548–4560. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.S.; Ahn, Y.J.; Lee, S.; Park, D.J.; Park, J.; Ha, S.M.; Seo, Y.J. Role of Kir4.1 Channels in Aminoglycoside-Induced Ototoxicity of Hair Cells. BioMed Res. Int. 2023, 2023, 4191999. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fracaro, S.; Hellies, F.; Marioni, G.; Brotto, D.; Franchella, S.; Zanoletti, E.; Albertin, G.; Astolfi, L. Role of Kir4.1 Channel in Auditory Function: Impact on Endocochlear Potential and Hearing Loss. Appl. Sci. 2024, 14, 4985. https://doi.org/10.3390/app14124985
Fracaro S, Hellies F, Marioni G, Brotto D, Franchella S, Zanoletti E, Albertin G, Astolfi L. Role of Kir4.1 Channel in Auditory Function: Impact on Endocochlear Potential and Hearing Loss. Applied Sciences. 2024; 14(12):4985. https://doi.org/10.3390/app14124985
Chicago/Turabian StyleFracaro, Silvia, Filippo Hellies, Gino Marioni, Davide Brotto, Sebastiano Franchella, Elisabetta Zanoletti, Giovanna Albertin, and Laura Astolfi. 2024. "Role of Kir4.1 Channel in Auditory Function: Impact on Endocochlear Potential and Hearing Loss" Applied Sciences 14, no. 12: 4985. https://doi.org/10.3390/app14124985