
Glycine-Conjugated α-Mangostins as Potential Estrogen Receptor Alpha (ERα) Antagonists through Pharmacophore Modeling, Docking Analysis, and Molecular Dynamics Simulations

 , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
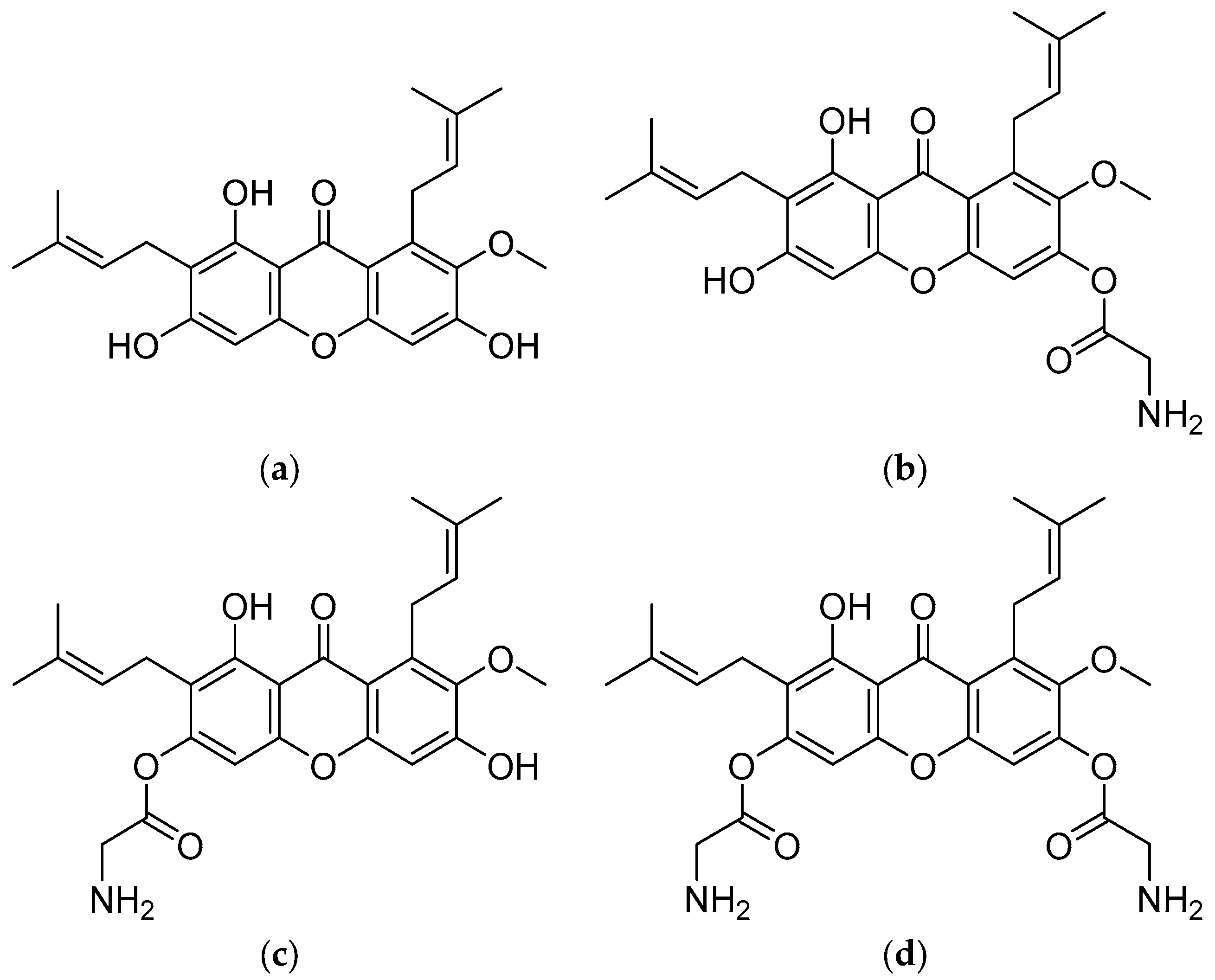
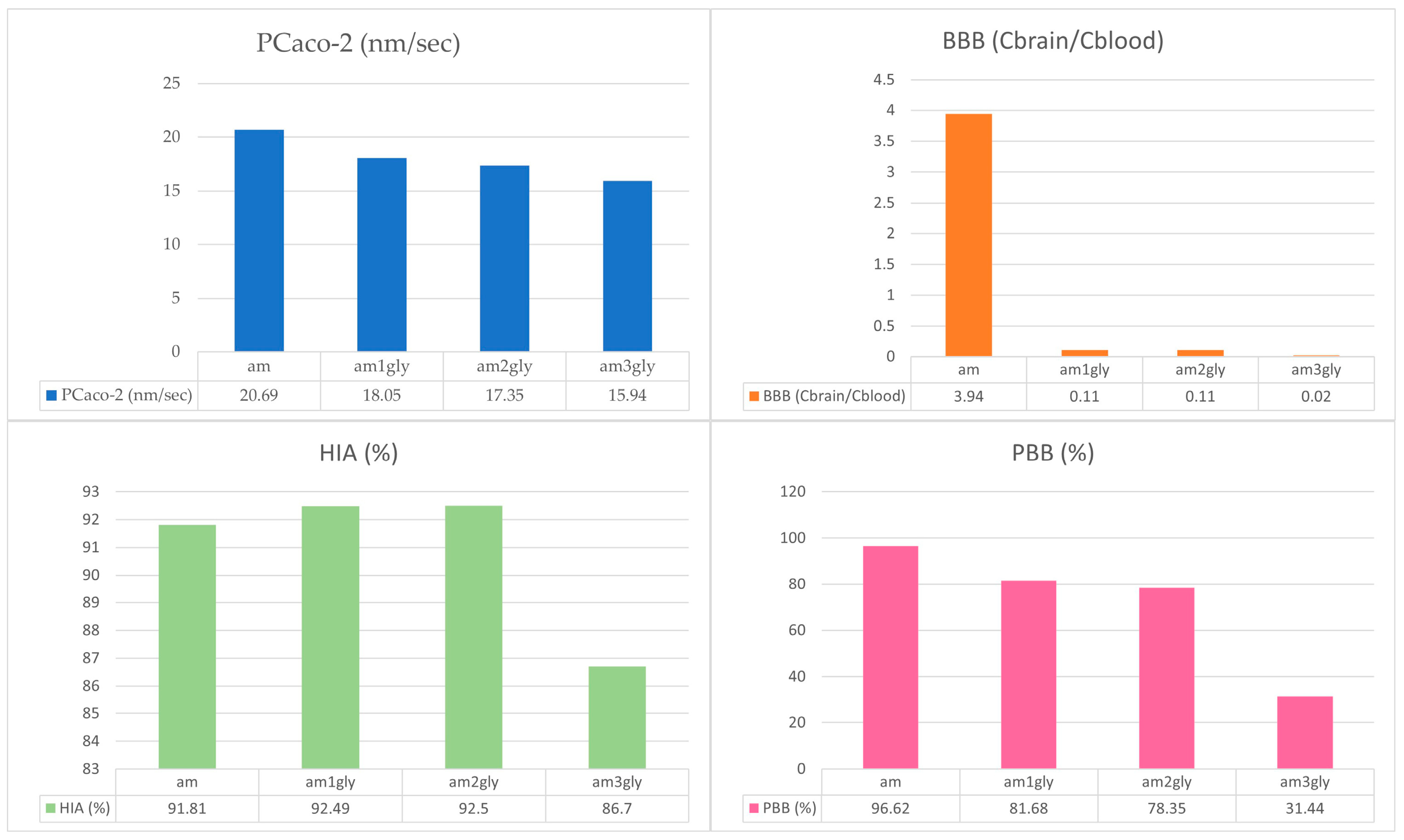
3.1. Pharmacokinetic and Toxicology Prediction
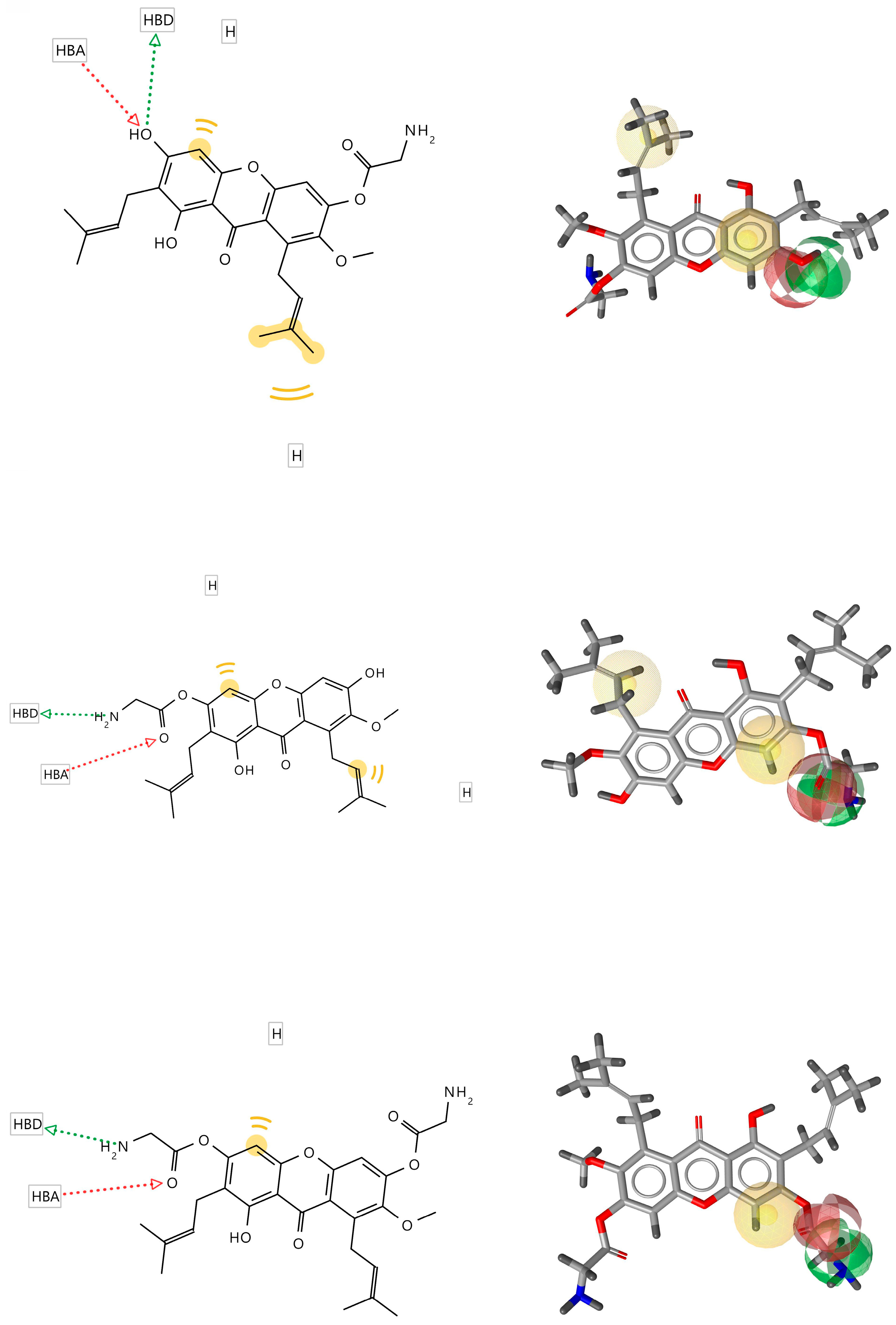
3.2. Pharmacophore Modeling
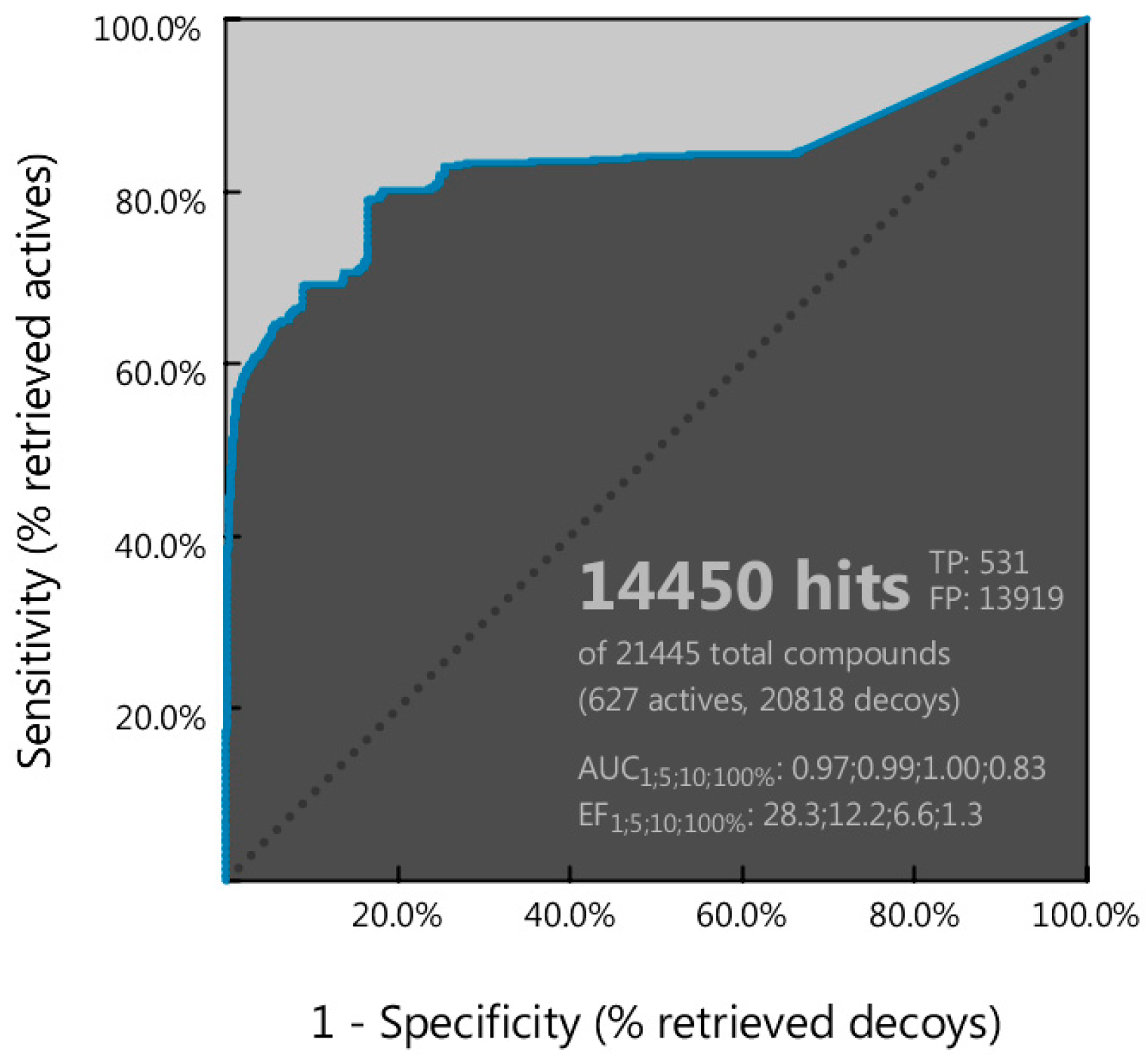
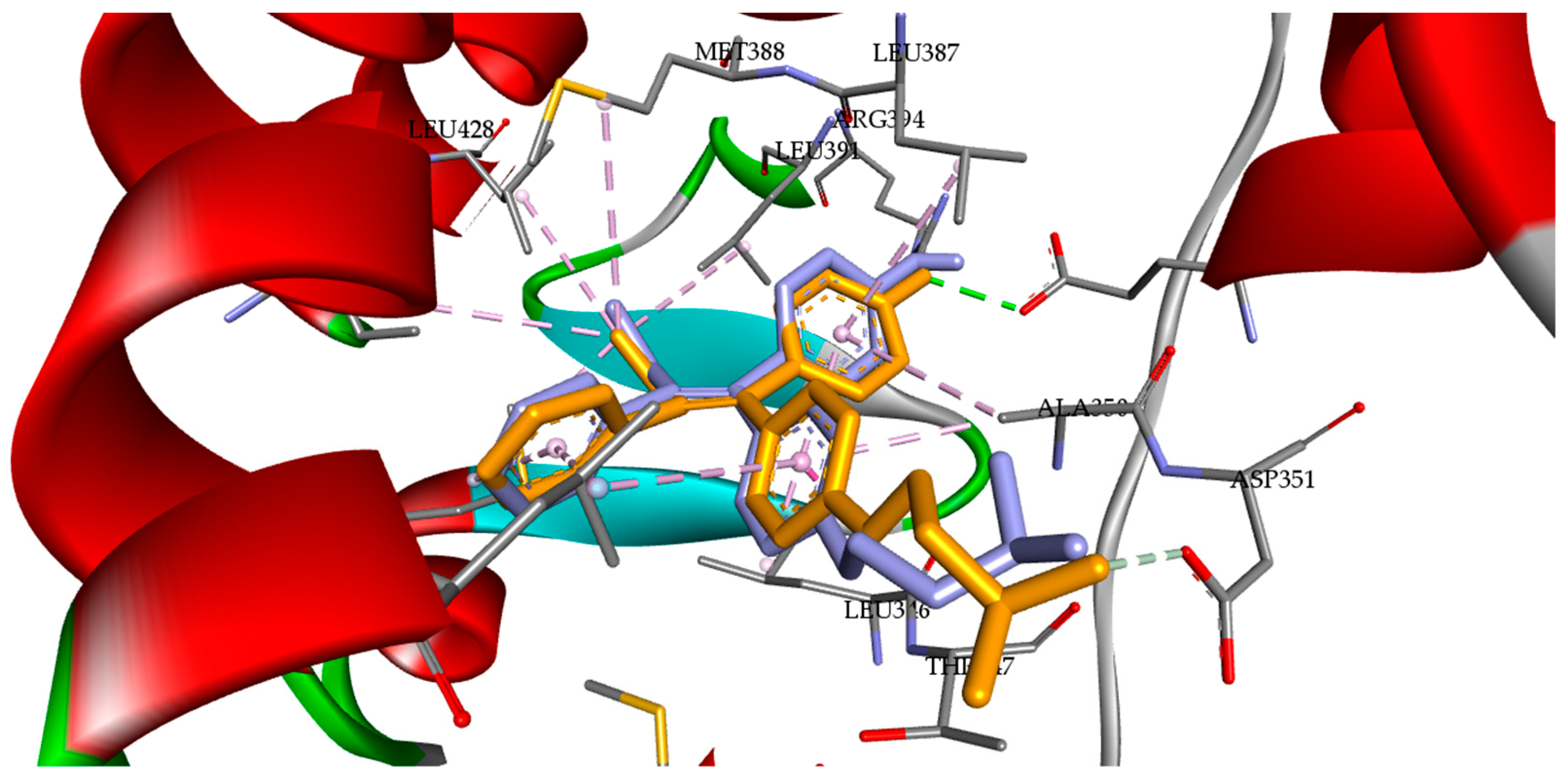
3.3. Molecular Docking
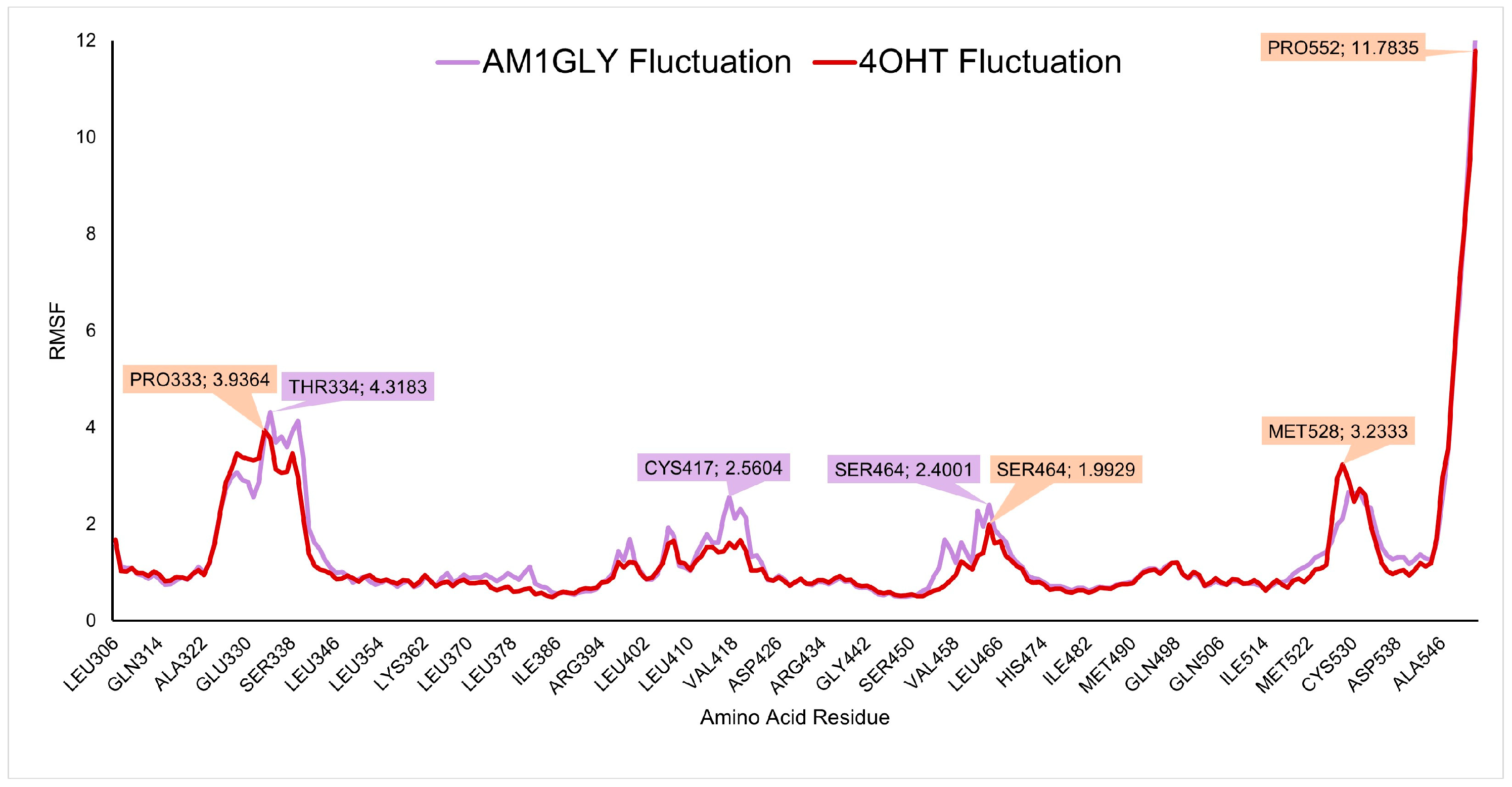
3.4. Molecular Dynamics Simulation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singla, P.; Salunke, D.B. Recent advances in steroid amino acid conjugates: Old scaffolds with new dimensions. Eur. J. Med. Chem. 2020, 187, 111909. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorganic. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, R.; De Vries, P.; Tulinsky, J.; Bellamy, G.; Baker, B.; Singer, J.W.; Klein, P. Synthesis and in vivo antitumor activity of poly(L-glutamic acid) conjugates of 20(S)-camptothecin. J. Med. Chem. 2003, 46, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.R.; Xu, B.; Yang, Y.Q.; Zhang, X.Y.; Fang, K.; Ma, T.; Wang, H.; Xue, N.N.; Chen, M.; Guo, W.B.; et al. Synthesis and biological evaluation of podophyllotoxin derivatives as selective antitumor agents. Eur. J. Med. Chem. 2018, 155, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Dubey, S.K.; Sharma, A.K.; Narain, U.; Misra, K.; Pati, U. Design, synthesis and characterization of some bioactive conjugates of curcumin with glycine, glutamic acid, valine and demethylenated piperic acid and study of their antimicrobial and antiproliferative properties. Eur. J. Med. Chem. 2008, 43, 1837–1846. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Wu, G.R.; Cai, D.S.; Xu, B.; Yan, M.M.; Ma, T.; Guo, W.B.; Zhang, W.X.; Huang, X.M.; Jia, X.h.; et al. Synthesis and biological activity of glycyrrhetinic acid derivatives as antitumor agents. Eur. J. Med. Chem. 2019, 178, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Li, Y.; Wang, W.; Deng, L. Bioactivity and pharmacological properties of α-mangostin from the mangosteen fruit: A review. Expert Opin. Ther. Patents 2018, 28, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Setiawati, A.; Riswanto, F.O.D.; Yuliani, S.H.; Istyastono, E.P. Anticancer Activity of Mangosteen Pericarp Dry Extract Against Mcf-7 Breast Cancer Cell Line Through Estrogen Receptor-α. Indones. J. Pharm. 2014, 25, 119. [Google Scholar] [CrossRef]
- Muchtaridi, M.; Wijaya, C.A. Anticancer potential of α-mangostin. Asian J. Pharm. Clin. Res. 2017, 10, 440–445. [Google Scholar] [CrossRef]
- Park, S.Y.; Lee, J.H.; Ko, S.Y.; Kim, N.; Kim, S.Y.; Lee, J.C. Antimicrobial activity of α-mangostin against Staphylococcus species from companion animals in vitro and therapeutic potential of α-mangostin in skin diseases caused by S. pseudintermedius. Front. Cell. Infect. Microbiol. 2023, 13, 1203663. [Google Scholar] [CrossRef]
- Herrera-Aco, D.R.; Medina-Campos, O.N.; Pedraza-Chaverri, J.; Sciutto-Conde, E.; Rosas-Salgado, G.; Fragoso-González, G. Alpha-mangostin: Anti-inflammatory and antioxidant effects on established collagen-induced arthritis in DBA/1J mice. Food Chem. Toxicol. 2019, 124, 300–315. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Li, J.; Ning, H.; Yuan, Z.; Zhong, Y.; Wu, S.; Zeng, J.-Z. α-Mangostin Induces Apoptosis and Inhibits Metastasis of Breast Cancer Cells via Regulating RXRα-AKT Signaling Pathway. Front. Pharmacol. 2021, 12, 739658. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.-J.; Gu, Q.-L.; Yang, K.; Ming, X.-J.; Wang, J.-X. Anticarcinogenic Effects of α-Mangostin: A Review. Planta Med. 2017, 83, 188–202. [Google Scholar] [CrossRef] [PubMed]
- Howell, A.; Howell, S.J. Tamoxifen evolution. Br. J. Cancer 2023, 128, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Decensi, A.; Maisonneuve, P.; Rotmensz, N.; Bettega, D.; Costa, A.; Sacchini, V.; Salvioni, A.; Travaglini, R.; Oliviero, P.; D’Aiuto, G.; et al. Effect of Tamoxifen on Venous Thromboembolic Events in a Breast Cancer Prevention Trial. Circulation 2005, 111, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Gong, C.; Xia, Y.; Zhou, Y.; Ye, T.; Mei, T.; Chen, H.; Chen, J. α-Mangostin Suppresses Melanoma Growth, Migration, and Invasion and Potentiates the Anti-tumor Effect of Chemotherapy. Int. J. Med. Sci. 2023, 20, 1220–1234. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Brunner, I.; Han, A.R.; Hamburger, M.; Kinghorn, A.D.; Frye, R.; Butterweck, V. Pharmacokinetics of α-mangostin in rats after intravenous and oral application. Mol. Nutr. Food Res. 2011, 55, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Pocasap, P.; Weerapreeyakul, N.; Timonen, J.; Järvinen, J.; Leppänen, J.; Kärkkäinen, J.; Rautio, J. Tyrosine–Chlorambucil Conjugates Facilitate Cellular Uptake through L-Type Amino Acid Transporter 1 (LAT1) in Human Breast Cancer Cell Line MCF-7. Int. J. Mol. Sci. 2020, 21, 2132. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, L.; Pan, J. The role of L-type amino acid transporter 1 in human tumors. Intractable Rare Dis. Res. 2015, 4, 165–169. [Google Scholar] [CrossRef]
- Koh, J.J.; Lin, S.; Aung, T.T.; Lim, F.; Zou, H.; Bai, Y.; Li, J.; Lin, H.; Pang, L.M.; Koh, W.L.; et al. Amino acid modified xanthone derivatives: Novel, highly promising membrane-active antimicrobials for multidrug-resistant gram-positive bacterial infections. J. Med. Chem. 2015, 58, 739–752. [Google Scholar] [CrossRef]
- Lumachi, F.; Santeufemia, D.A.; Basso, S.M. Current medical treatment of estrogen receptor-positive breast cancer. World J. Biol. Chem. 2015, 6, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Saha, T.; Makar, S.; Swetha, R.; Gutti, G.; Singh, S.K. Estrogen signaling: An emanating therapeutic target for breast cancer treatment. Eur. J. Med. Chem. 2019, 177, 116–143. [Google Scholar] [CrossRef] [PubMed]
- Maximov, P.Y.; Fan, P.; Abderrahman, B.; Curpan, R.; Jordan, V.C. Estrogen Receptor Complex to Trigger or Delay Estrogen-Induced Apoptosis in Long-Term Estrogen Deprived Breast Cancer. Front. Endocrinol. 2022, 13, 869562. [Google Scholar] [CrossRef] [PubMed]
- Csizmadia, P. MarvinSketch and MarvinView: Molecule Applets for the World Wide Web. In Proceedings of the 3rd International Electronic Conference on Synthetic Organic Chemistry; 1–30 November 1999; MDPI: Basel, Switzerland, 1999. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeerschd, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- García-Rodríguez, A.; Vila, L.; Cortés, C.; Hernández, A.; Marcos, R. Exploring the usefulness of the complex in vitro intestinal epithelial model Caco-2/HT29/Raji-B in nanotoxicology. Food Chem. Toxicol. 2018, 113, 162–170. [Google Scholar] [CrossRef]
- Maliehe, T.; Tsilo, P.; Shandu, J.; Shandu, J. Computational Evaluation of ADMET Properties and Bioactive Score of Compounds from Encephalartos ferox. Pharmacogn. J. 2020, 12, 1357–1362. [Google Scholar] [CrossRef]
- Nisha, C.M.; Kumar, A.; Vimal, A.; Bai, B.M.; Pal, D.; Kumar, A. Docking and ADMET prediction of few GSK-3 inhibitors divulges 6-bromoindirubin-3-oxime as a potential inhibitor. J. Mol. Graph. Model. 2016, 65, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Batista, M.A.; de Lima Teixeira dos Santos, A.V.T.; do Nascimento, A.L.; Moreira, L.F.; Souza, I.R.S.; da Silva, H.R.; Pereira, A.C.M.; da Silva Hage-Melim, L.I.; Carvalho, J.C.T. Potential of the Compounds from Bixa orellana Purified Annatto Oil and Its Granules (Chronic®) against Dyslipidemia and Inflammatory Diseases: In Silico Studies with Geranylgeraniol and Tocotrienols. Molecules 2022, 27, 1584. [Google Scholar] [CrossRef]
- Barros, B.; Oliveira, M.; Morais, S. Unveiling Urinary Mutagenicity by the Ames Test for Occupational Risk Assessment: A Systematic Review. Int. J. Environ. Res. Public Health 2022, 19, 13074. [Google Scholar] [CrossRef]
- Pradeepkiran, J.A.; Sainath, S.B.; Shrikanya, K.V.L. In silico validation and ADMET analysis for the best lead molecules. In Brucella Melitensis; Elsevier: Amsterdam, The Netherlands, 2021; pp. 133–176. [Google Scholar]
- Bahamonde, J.; Brenseke, B.; Chan, M.Y.; Kent, R.D.; Vikesland, P.J.; Prater, M.R. Gold Nanoparticle Toxicity in Mice and Rats: Species Differences. Toxicol. Pathol. 2018, 46, 431–443. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2004, 45, 160–169. [Google Scholar] [CrossRef]
- Yoshimori, A.; Kawasaki, E.; Kanai, C.; Tasaka, T. Strategies for Design of Molecular Structures with a Desired Pharmacophore Using Deep Reinforcement Learning. Chem. Pharm. Bull. 2020, 68, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998, 95, 927–937. [Google Scholar] [CrossRef]
- Muchtaridi, M.; Yusuf, M.; Diantini, A.; Choi, S.B.; Al-Najjar, B.O.; Manurung, J.V.; Subarnas, A.; Achmad, T.H.; Wardhani, S.R.; Wahab, H.A. Potential activity of fevicordin-A from Phaleria macrocarpa (Scheff) Boerl. seeds as estrogen receptor antagonist based on cytotoxicity and molecular modelling studies. Int. J. Mol. Sci. 2014, 15, 7225–7249. [Google Scholar] [CrossRef] [PubMed]
- El-Hachem, N.; Haibe-Kains, B.; Khalil, A.; Kobeissy, F.H.; Nemer, G. AutoDock and AutoDockTools for Protein-Ligand Docking: Beta-Site Amyloid Precursor Protein Cleaving Enzyme 1(BACE1) as a Case Study. In Neuroproteomics: Methods and Protocols; Kobeissy, F.H., Stevens, J.S.M., Eds.; Springer: New York, NY, USA, 2017; pp. 391–403. [Google Scholar]
- Hou, X.; Du, J.; Zhang, J.; Du, L.; Fang, H.; Li, M. How to Improve Docking Accuracy of AutoDock4.2: A Case Study Using Different Electrostatic Potentials. J. Chem. Inf. Model. 2013, 53, 188–200. [Google Scholar] [CrossRef]
- Setyawati, L.U.; Parlan, F.I.H.B.; Ikram, N.K.K.; Yusuf, M.; Muchtaridi, M. Molecular Dynamic Simulation and 3d-pharmacophore Modeling of Alpha Mangostin and Its Derivatives against Estrogen Alpha Receptor. Lett. Drug Des. Discov. 2024, 21, 1103–1119. [Google Scholar] [CrossRef]
- Muchtaridi, M.; Syahidah, H.N.; Subarnas, A.; Yusuf, M.; Bryant, S.D.; Langer, T. Molecular docking and 3D-pharmacophore modeling to study the interactions of chalcone derivatives with estrogen receptor alpha. Pharmaceuticals 2017, 10, 81. [Google Scholar] [CrossRef]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef]
- Kumari, A.; Singh, A.; Raghava, M.; Singh, M.; Tiwari, P.K.; Prakash, S.; Kumar, A.; Bansal, P. An approach of computer-aided drug design (CADD) tools for in silico assessment of various inhibitors of lanosterol-14α demethylase. Mater. Today Proc. 2023. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.; Brozell, S.R.; Cerutti, D.S.; Cheatham Iii, T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E. Amber 2021; University of California: San Francisco, CA, USA, 2021. [Google Scholar]
- Sinyani, A.; Idowu, K.; Shunmugam, L.; Kumalo, H.M.; Khan, R. A molecular dynamics perspective into estrogen receptor inhibition by selective flavonoids as alternative therapeutic options. J. Biomol. Struct. Dyn. 2023, 41, 4093–4105. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.; Xu, Y.; Shi, Y.; Yu, Q.; Liu, J.; Xu, L. Discovery of novel natural compound inhibitors targeting estrogen receptor α by an integrated virtual screening strategy. J. Mol. Model. 2019, 25, 278. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Dixit, A.; Kashaw, S.K. Ligand and structure based virtual screening of chemical databases to explore potent small molecule inhibitors against breast invasive carcinoma using recent computational technologies. J. Mol. Graph. Model. 2020, 98, 107591. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Konishi, K.; Yamazaki, Y.; Taki, Y.; Sakane, T.; Sezaki, H.; Furuyama, Y. New and better protocols for a short-term Caco-2 cell culture system. J. Pharm. Sci. 2002, 91, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, P. Advances in Pharmacophore Modeling and Its Role in Drug Designing. In Computer-Aided Drug Design; Singh, D.B., Ed.; Springer: Singapore, 2020; pp. 223–243. [Google Scholar]
- Tran-Nguyen, V.K.; Da Silva, F.; Bret, G.; Rognan, D. All in One: Cavity Detection, Druggability Estimate, Cavity-Based Pharmacophore Perception, and Virtual Screening. J. Chem. Inf. Model. 2019, 59, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Tu, Y.; Ågren, H.; Eriksson, L.A. Characterization of agonist binding to His524 in the estrogen receptor α ligand binding domain. J. Phys. Chem. B 2012, 116, 4823–4830. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, S.A.; Fatima, N. In silico analysis and molecular docking studies of potential angiotensin-converting enzyme inhibitor using quercetin glycosides. Pharmacogn. Mag. 2015, 11, S123. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Shu, X.; Zhou, H.; Beckford, F.A.; Misir, M. Algorithm selection for protein–ligand docking: Strategies and analysis on ACE. Sci. Rep. 2023, 13, 8219. [Google Scholar] [CrossRef] [PubMed]
- Katoch, S.; Chauhan, S.S.; Kumar, V. A review on genetic algorithm: Past, present, and future. Multimed. Tools Appl. 2021, 80, 8091–8126. [Google Scholar] [CrossRef]
- Shylaja, R.; Loganathan, C.; Kabilan, S.; Vijayakumar, T.; Meganathan, C. Synthesis and evaluation of the antagonistic activity of 3-acetyl-2H-benzo[g]chromen-2-one against mutant Y537S estrogen receptor alpha via E-Pharmacophore modeling, molecular docking, molecular dynamics, and in-vitro cytotoxicity studies. J. Mol. Struct. 2021, 1224, 129289. [Google Scholar] [CrossRef]
- Kaur, D.; Choudhury, C.; Yadav, R.; Kumari, L.; Bhatia, A. Aspirin as a potential drug repurposing candidate targeting estrogen receptor alpha in breast cancer: A molecular dynamics and in-vitro study. J. Biomol. Struct. Dyn. 2024, 27, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Muchtaridi, M.; Megantara, S.; Dermawan, D.; Yusuf, M. Antagonistic mechanism of α-mangostin derivatives against human estrogen receptor α of breast cancer using molecular dynamics simulation. Rasayan J. Chem. 2019, 12, 1927–1934. [Google Scholar] [CrossRef]
- Ishola, A.A.; Joshi, T.; Abdulai, S.I.; Tijjani, H.; Pundir, H.; Chandra, S. Molecular basis for the repurposing of histamine H2-receptor antagonist to treat COVID-19. J. Biomol. Struct. Dyn. 2022, 40, 5785–5802. [Google Scholar] [CrossRef]
- Mardianingrum, R.; Yusuf, M.; Hariono, M.; Mohd Gazzali, A.; Muchtaridi, M. α-Mangostin and its derivatives against estrogen receptor alpha. J. Biomol. Struct. Dyn. 2022, 40, 2621–2634. [Google Scholar] [CrossRef] [PubMed]
- Lohachova, K.O.; Kyrychenko, A.; Kalugin, O.N. Critical assessment of popular biomolecular force fields for molecular dynamics simulations of folding and enzymatic activity of main protease of coronavirus SARS-CoV-2. Biophys. Chem. 2024, 311, 107258. [Google Scholar] [CrossRef]
- Muhammad, S.; Saba, A.; Khera, R.A.; Al-Sehemi, A.G.; Algarni, H.; Iqbal, J.; Alshahrani, M.Y.; Chaudhry, A.R. Virtual screening of potential inhibitor against breast cancer-causing estrogen receptor alpha (ERα): Molecular docking and dynamic simulations. Mol. Simul. 2022, 48, 1163–1174. [Google Scholar] [CrossRef]
- Alaqarbeh, M.; El Mchichi, L.; Abouzied, A.S.; Bouzzine, S.M.; Huwaimel, B.; Bouachrine, M. Computational investigation of structural-biological inhibitory activity for Au (III) porphyrin complexes against MCF-7 human breast cancer. Chem. Data Collect. 2023, 48, 101094. [Google Scholar] [CrossRef]
- Muhammad, S.; Zahir, N.; Bibi, S.; Alshahrani, M.Y.; Chaudhry, A.R.; Sarwar, F.; Tousif, M.I. Computational prediction for designing novel ketonic derivatives as potential inhibitors for breast cancer: A trade-off between drug likeness and inhibition potency. Comput. Biol. Chem. 2024, 109, 108020. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, C.; Lu, J.; Wu, J.; Li, C.; Hu, Z.; Tian, W.; Yang, L.; Xiang, J.; Zhou, H.; et al. Sesterterpene MHO7 suppresses breast cancer cells as a novel estrogen receptor degrader. Pharmacol. Res. 2019, 146, 104294. [Google Scholar] [CrossRef] [PubMed]
- Geetha Rani, Y.; Lakshmi, B.S. Structural insight into the antagonistic action of diarylheptanoid on human estrogen receptor alpha. J. Biomol. Struct. Dyn. 2019, 37, 1189–1203. [Google Scholar] [CrossRef]
- Amneh, S.; Rohana, A.; Melati, K.; Shikin Faezah, S. Investigation of Newly Designed Human Estrogen Receptor Inhibitors from Benzophenones Derivatives (BPs) by Molecular Docking and Molecular Dynamic Simulation. Aust. J. Basic Appl. Sci. 2016, 10, 49–59. [Google Scholar]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Roa, C.; Cortes, E.; Cuesta, S.A.; Mora, J.R.; Paz, J.L.; Flores-Sumoza, M.; Márquez, E.A. Study of potential inhibition of the estrogen receptor α by cannabinoids using an in silico approach: Agonist vs antagonist mechanism. Comput. Biol. Med. 2023, 152, 106403. [Google Scholar] [CrossRef] [PubMed]
- Ananth, A.H.; Manikandan, N.; Rajan, R.K.; Elancheran, R.; Lakshmithendral, K.; Ramanathan, M.; Bhattacharjee, A.; Kabilan, S. Design, Synthesis, and Biological Evaluation of 2-(2-Bromo-3-nitrophenyl)-5-phenyl-1, 3, 4-oxadiazole Derivatives as Possible Anti-Breast Cancer Agents. Chem. Biodivers. 2020, 17, e1900659. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecules | Pharmacokinetics Prediction | Toxicity Prediction | |||||
|---|---|---|---|---|---|---|---|
| PCaco-2 (nm/s) | BBB (Cbrain/Cblood) | HIA (%) | PBB (%) | Rat | Mice | Ames | |
| am | 20.69 | 3.94 | 91.81 | 96.62 | + | - | - |
| Am1Gly | 18.05 | 0.11 | 92.49 | 81.68 | - | + | - |
| Am2Gly | 17.35 | 0.11 | 92.5 | 78.35 | - | - | - |
| Am3Gly | 15.94 | 0.02 | 86.7 | 31.44 | - | - | - |
| Compounds | Binding Affinity (kcal/mol) | Molecular Interactions | |
|---|---|---|---|
| Hydrogen Bond | Alkyl | ||
| 4OHT | −11.66 | Arg394, Glu353 | Leu346, Leu391, Met421, Leu387, Ala350, Leu525 |
| Am1Gly | −10.91 | Glu353, Asp351, Thr347 | Leu346, Leu525, Met421, Ile424, Ala350, Leu525 |
| Am2Gly | −10.41 | Asp351, Thr347 | Leu346, Leu525, Met421, Ile424, Ala350, Leu384 |
| System | H-Bond Acceptor (atom@res) | H-Bond Donor (atom@res) | Occupancy (%) | Avg. Distance (Å) | Avg. Angle (°) |
|---|---|---|---|---|---|
| 4OHT | O@Glu353 | H@4OHT | 10.38 | 2.65 | 160.57 |
| O@Glu353 | H@4OHT | 9.86 | 2.66 | 158.39 | |
| N@4OHT | H@Ser527 | 8.53 | 2.83 | 164.10 | |
| O@Leu387 | H@4OHT | 7.19 | 2.78 | 164.08 | |
| O@4OHT | H@Arg394 | 2.02 | 2.91 | 147.06 | |
| O@4OHT | H@Ser527 | 1.51 | 2.84 | 155.07 | |
| O@Phe404 | H@4OHT | 0.59 | 2.79 | 152.73 | |
| N@4OHT | H@Lys529 | 0.20 | 2.88 | 156.65 | |
| N@4OHT | H@Lys529 | 0.19 | 2.89 | 158.56 | |
| N@4OHT | H@Lys529 | 0.17 | 2.87 | 153.03 | |
| O@4OHT | H@Arg394 | 0.13 | 2.91 | 143.01 | |
| O@4OHT | H@Lys529 | 0.11 | 2.89 | 149.94 | |
| O@4OHT | H@Thr347 | 0.11 | 2.84 | 161.15 | |
| O@4OHT | H@Lys529 | 0.09 | 2.89 | 151.44 | |
| O@Leu349 | H@4OHT | 0.07 | 2.77 | 164.23 | |
| Am1Gly | O@Asp351 | H@Am1Gly | 49.80 | 2.60 | 164.41 |
| O@Phe404 | H@Am1Gly | 12.85 | 2.87 | 155.19 | |
| O@Glu353 | H@Am1Gly | 4.87 | 2.85 | 160.29 | |
| H@Am1Gly | H@Arg394 | 3.57 | 2.86 | 147.49 | |
| H@Am1Gly | H@Asn532 | 0.61 | 2.92 | 159.48 | |
| N@Am1Gly | H@Arg394 | 0.12 | 2.90 | 142.21 | |
| O@Asn532 | H@Am1Gly | 0.12 | 2.80 | 144.27 | |
| N@Am1Gly | H@Arg394 | 0.12 | 2.90 | 149.09 | |
| O@Am1Gly | H@Leu525 | 0.11 | 2.88 | 158.21 | |
| O@Am1Gly | H@Lys531 | 0.06 | 2.91 | 151.57 | |
| O@Am1Gly | H@Arg394 | 0.05 | 2.88 | 141.58 | |
| O@Am1Gly | H@Lys531 | 0.05 | 2.90 | 150.17 | |
| O@Am1Gly | H@Lys531 | 0.05 | 2.91 | 150.97 | |
| O@Am1Gly | H@Lys531 | 0.03 | 2.87 | 149.78 | |
| O@Am1Gly | H@Asn532 | 0.03 | 2.91 | 147.49 | |
| O@Am1Gly | H@Lys531 | 0.03 | 2.91 | 155.83 | |
| O@Thr347 | H@Am1Gly | 0.03 | 2.80 | 157.33 | |
| O@Am1Gly | H@Lys531 | 0.02 | 2.92 | 151.21 | |
| O@Leu349 | H@Am1Gly | 0.02 | 2.90 | 151.34 | |
| O@Leu349 | H@Am1Gly | 0.01 | 2.90 | 146.00 | |
| O@Am1Gly | H@Lys531 | 0.01 | 2.91 | 153.19 | |
| N@Arg394 | H@Am1Gly | 0.01 | 2.91 | 150.45 | |
| N@Arg394 | H@Am1Gly | 0.01 | 2.95 | 141.14 | |
| O@Leu346 | H@Am1Gly | 0.01 | 2.84 | 152.03 | |
| O@Am1Gly | H@Arg394 | 0.01 | 2.88 | 151.99 | |
| O@Thr347 | H@Am1Gly | 0.01 | 2.76 | 149.76 | |
| O@Am1Gly | H@Lys531 | 0.01 | 2.87 | 150.60 |
| Energy Component | Bond Energy (kcal/mol) | |
|---|---|---|
| 4OHT | Am1Gly | |
| ΔGVDW | −53.14 | −56.34 |
| ΔGEL | −10.45 | −26.49 |
| ΔEGB | 17.87 | 42.11 |
| ΔESURF | −7.53 | −8.07 |
| ΔGGAS | −63.59 | −82.23 |
| ΔGSOLV | 10.34 | 32.04 |
| ΔGTOTAL | −53.25 | −48.79 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arifian, H.; Maharani, R.; Megantara, S.; Ikram, N.K.K.; Muchtaridi, M. Glycine-Conjugated α-Mangostins as Potential Estrogen Receptor Alpha (ERα) Antagonists through Pharmacophore Modeling, Docking Analysis, and Molecular Dynamics Simulations. Appl. Sci. 2024, 14, 5549. https://doi.org/10.3390/app14135549
Arifian H, Maharani R, Megantara S, Ikram NKK, Muchtaridi M. Glycine-Conjugated α-Mangostins as Potential Estrogen Receptor Alpha (ERα) Antagonists through Pharmacophore Modeling, Docking Analysis, and Molecular Dynamics Simulations. Applied Sciences. 2024; 14(13):5549. https://doi.org/10.3390/app14135549
Chicago/Turabian StyleArifian, Hanggara, Rani Maharani, Sandra Megantara, Nur Kusaira Khairul Ikram, and Muchtaridi Muchtaridi. 2024. "Glycine-Conjugated α-Mangostins as Potential Estrogen Receptor Alpha (ERα) Antagonists through Pharmacophore Modeling, Docking Analysis, and Molecular Dynamics Simulations" Applied Sciences 14, no. 13: 5549. https://doi.org/10.3390/app14135549
APA StyleArifian, H., Maharani, R., Megantara, S., Ikram, N. K. K., & Muchtaridi, M. (2024). Glycine-Conjugated α-Mangostins as Potential Estrogen Receptor Alpha (ERα) Antagonists through Pharmacophore Modeling, Docking Analysis, and Molecular Dynamics Simulations. Applied Sciences, 14(13), 5549. https://doi.org/10.3390/app14135549