
Biosimilar Medicines: From Development Process to Marketing Authorization by the EMA and the FDA


Abstract
:1. Introduction
2. Overview of Biosimilar Medicines
2.1. Biological Medicines
2.2. Biosimilar Medicines
2.3. Intended Copies, Biobetters and Stand-Alone Biologics
2.4. Biosimilars vs. Generics
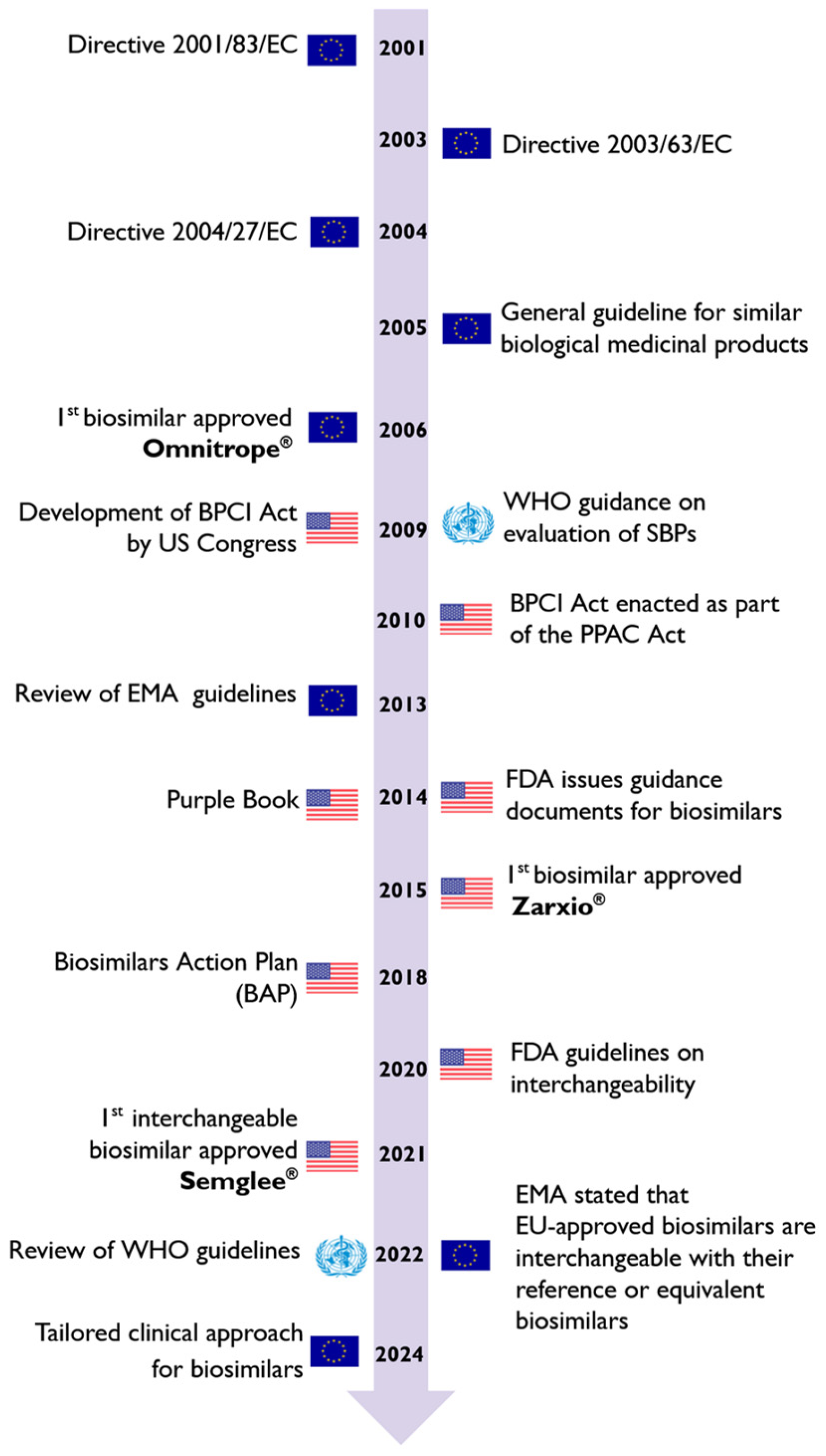
3. Biosimilar Medicines Regulatory Framework
4. Development of Biosimilar Medicines
4.1. Selection and Characterization of the Reference Product
4.2. Cell Line Creation
4.3. Cultivation and Production
4.4. Isolation and Purification
4.5. Formulation, Filling and Finishing
5. Demonstration of Biosimilarity
5.1. Quality Comparability Studies
Statistical Approaches to Evaluate Analytical Similarity
5.2. Non-Clinical Comparability Studies
5.3. Clinical Comparability Studies
5.3.1. Clinical Pharmacology Studies (PK/PD)
5.3.2. Efficacy Studies
5.3.3. Safety Evaluation
5.3.4. Immunogenicity Concerns
5.4. Other Relevant Aspects
5.4.1. Extrapolation of Indication
5.4.2. Nomenclature
5.4.3. Interchangeability
5.4.4. Patent Protection and Market Exclusivity
6. Biosimilar Medicines: Marketing Authorization
6.1. Data Requirements
6.2. European Medicines Agency and Food and Drug Administration Pathways
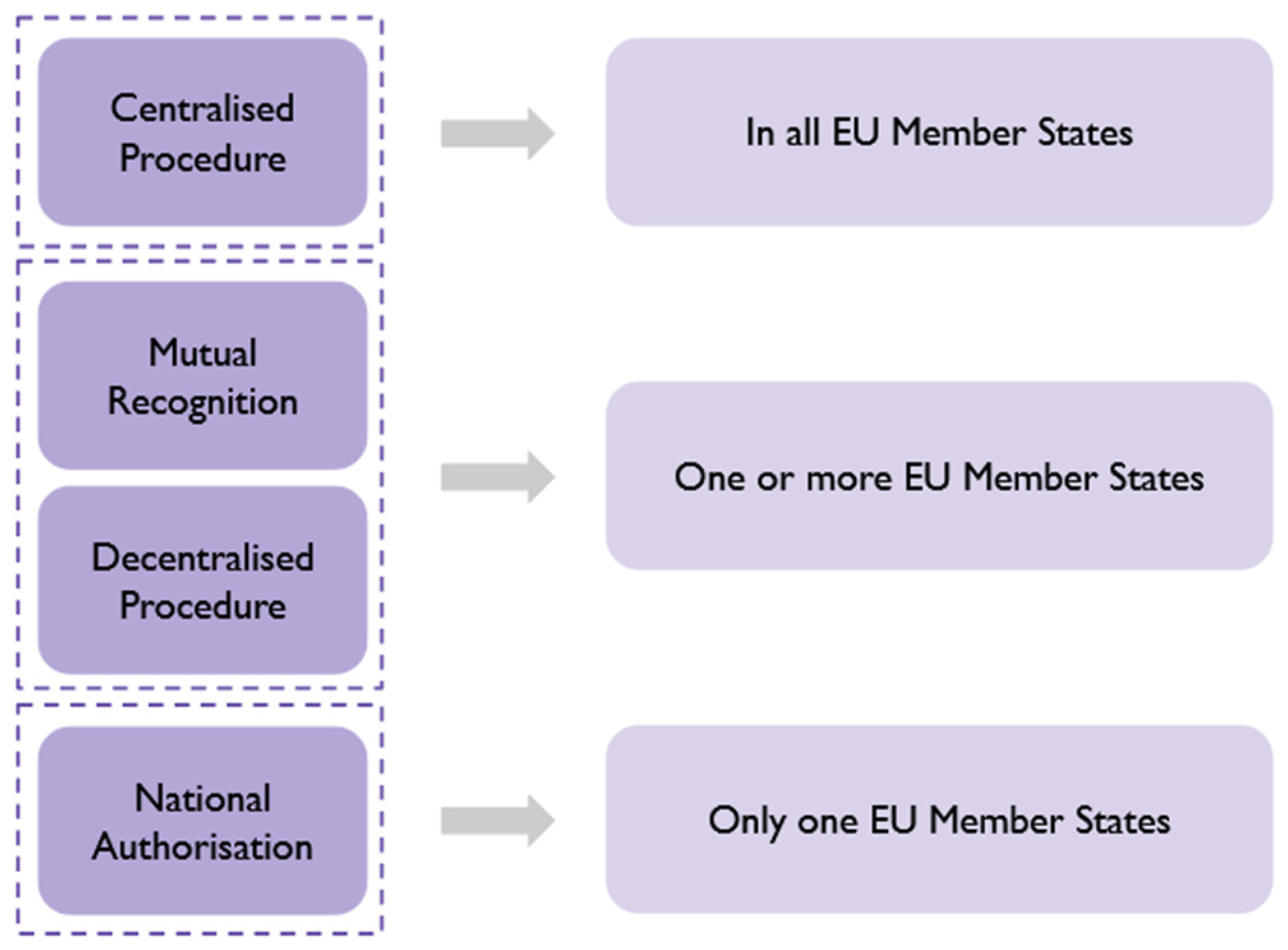
6.2.1. European Medicines Agency
6.2.2. Food and Drug Administration
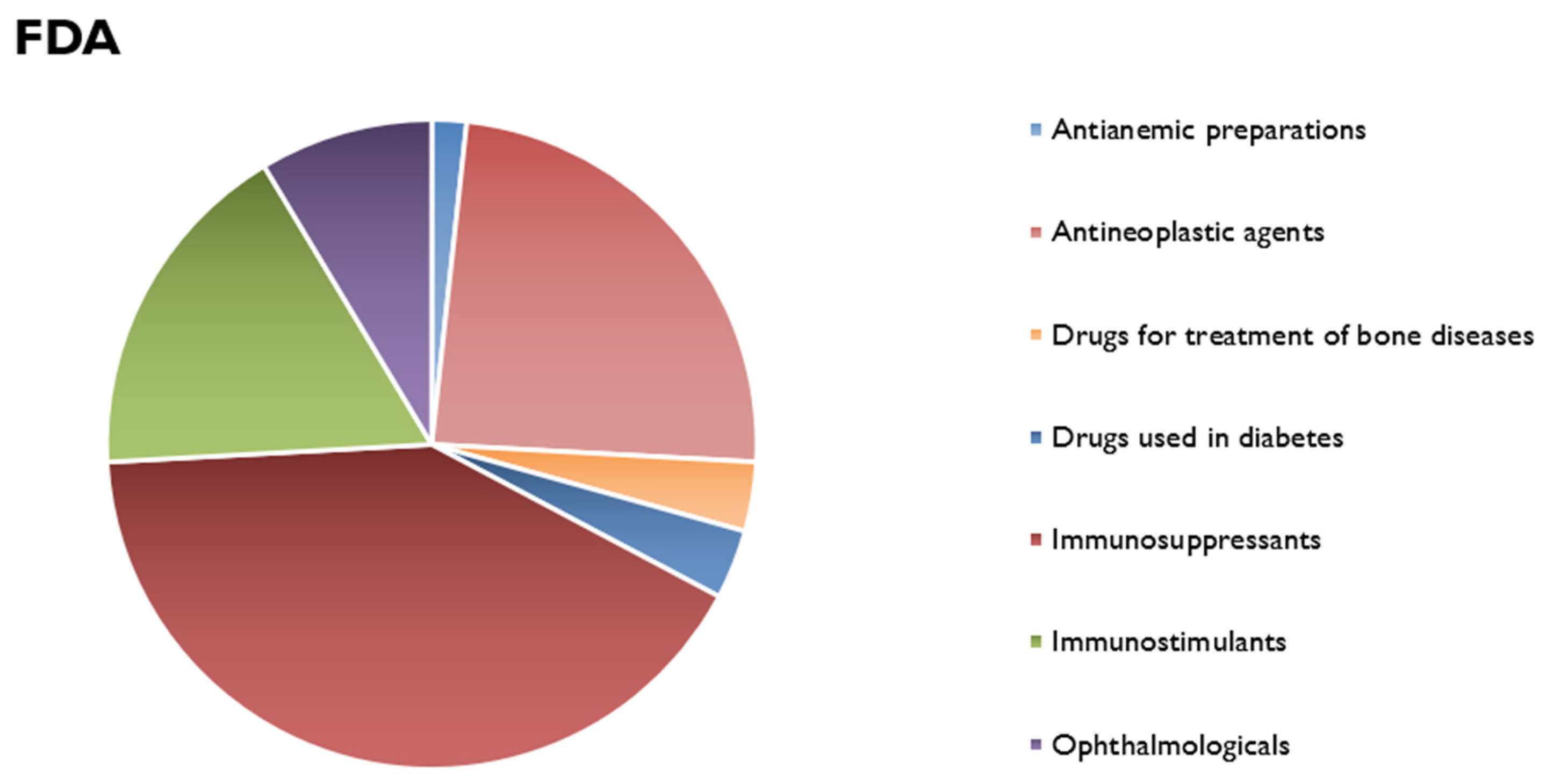
7. Market Evaluation
8. Conclusions and Future Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gherghescu, I.; Delgado-Charro, M.B. The Biosimilar Landscape: An Overview of Regulatory Approvals by the EMA and FDA. Pharmaceutics 2020, 13, 48. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency; European Commission. Biosimilars in the EU—Information Guide for Healthcare Professionals. 2019. Available online: https://www.ema.europa.eu/en/documents/leaflet/biosimilars-eu-information-guide-healthcare-professionals_en.pdf (accessed on 7 December 2023).
- Iskit, A.B. Key concepts in biosimilar medicines: What physicians must know. North. Clin. Istanb. 2022, 9, 86–91. [Google Scholar] [CrossRef]
- Simoens, S. Biosimilar Medicines and Cost-Effectiveness. ClinicoEcon. Outcomes Res. 2011, 3, 29–36. [Google Scholar] [CrossRef]
- Smith, S. Manufacturing, Approval and Advantages of Biosimilars. Edu J. Int. Aff. Res. 2022, 1, 30–38. [Google Scholar]
- Declerck, P.; Danesi, R.; Petersel, D.; Jacobs, I. The Language of Biosimilars: Clarification, Definitions, and Regulatory Aspects. Drugs 2017, 77, 671–677. [Google Scholar] [CrossRef]
- Schiestl, M.; Zabransky, M.; Sörgel, F. Ten Years of Biosimilars in Europe: Development and Evolution of the Regulatory Pathways. Drug Des. Dev. Ther. 2017, 11, 1509–1515. [Google Scholar] [CrossRef]
- Kulasekararaj, A.; Brodsky, R.; Kulagin, A.; Jang, J.H. Biosimilars in Rare Diseases: A Focus on Paroxysmal Nocturnal Hemoglobinuria. Haematologica 2023, 108, 1232–1243. [Google Scholar] [CrossRef] [PubMed]
- Mirjalili, S.Z.; Sabourian, R.; Sadeghalvad, M.; Rezaei, N. Therapeutic Applications of Biosimilar Monoclonal Antibodies: Systematic Review of the Efficacy, Safety, and Immunogenicity in Autoimmune Disorders. Int. Immunopharmacol. 2021, 101, 108305. [Google Scholar] [CrossRef]
- European Commission. What You Need to Know about Biosimilar Medicinal Products—A Consensus Information Document. 2013. Available online: https://ec.europa.eu/docsroom/documents/8242/ (accessed on 14 December 2023).
- Mitra, S.; Murthy, G.S. Bioreactor control systems in the biopharmaceutical industry: A critical perspective. Syst. Microbiol. Biomanuf. 2022, 2, 91–112. [Google Scholar] [CrossRef]
- Biological Product Definitions. Available online: https://www.fda.gov/files/drugs/published/Biological-Product-Definitions.pdf (accessed on 5 January 2024).
- Bachu, R.D.; Abou-Dahech, M.; Balaji, S.; Boddu, S.H.S.; Amos, S.; Singh, V.; Babu, R.J.; Tiwari, A.K. Oncology Biosimilars: New Developments and Future Directions. Cancer Rep. 2022, 5, e1720. [Google Scholar] [CrossRef]
- European Medicines Agency. Guideline on Similar Biological Medicinal Products. CHMP/437/04 Rev. 1. 2014. Available online: https://www.ema.europa.eu/system/files/documents/scientific-guideline/wc500176768_en.pdf (accessed on 8 January 2024).
- Kirchhoff, C.F.; Wang, X.Z.M.; Conlon, H.D.; Anderson, S.; Ryan, A.M.; Bose, A. Biosimilars: Key Regulatory Considerations and Similarity Assessment Tools. Biotechnol. Bioeng. 2017, 114, 2696–2705. [Google Scholar] [CrossRef] [PubMed]
- Agbogbo, F.K.; Ecker, D.M.; Farrand, A.; Han, K.; Khoury, A.; Martin, A.; McCool, J.; Rasche, U.; Rau, T.D.; Schmidt, D.; et al. Current Perspectives on Biosimilars. J. Ind. Microbiol. Biotechnol. 2019, 46, 1297–1311. [Google Scholar] [CrossRef]
- Tesser, J.R.; Furst, D.E.; Jacobs, I. Biosimilars and the Extrapolation of Indications for Inflammatory Conditions. Biol. Targets Ther. 2017, 11, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Dutta, B.; Huys, I.; Vulto, A.G.; Simoens, S. Identifying Key Benefits in European Off-Patent Biologics and Biosimilar Markets: It Is Not Only About Price! BioDrugs Clin. Immunother. Biopharm. Gene Ther. 2020, 34, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Gámez-Belmonte, R.; Hernández-Chirlaque, C.; Arredondo-Amador, M.; Aranda, C.J.; González, R.; Martínez-Augustin, O.; Medina, F.S. Biosimilars: Concepts and Controversies. Pharmacol. Res. 2018, 133, 251–264. [Google Scholar] [CrossRef]
- Kapur, M.; Nirula, S.; Naik, M.P. Future of Anti-VEGF: Biosimilars and Biobetters. Int. J. Retin. Vitr. 2022, 8, 2. [Google Scholar] [CrossRef]
- Camacho, L.H.; Frost, C.P.; Abella, E.; Morrow, P.K.; Whittaker, S. Biosimilars 101: Considerations for U.S. Oncologists in Clinical Practice. Cancer Med. 2014, 3, 889–899. [Google Scholar] [CrossRef]
- Van de Vooren, K.; Curto, A.; Garattini, L. Biosimilar versus Generic Drugs: Same but Different? Appl. Health Econ. Health Policy 2015, 13, 125–127. [Google Scholar] [CrossRef]
- European Medicines Agency. ICH Topic Q 5 E Comparability of Biotechnological/Biological Products CPMP/ICH/5721/03. 2005. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-5-e-comparability-biotechnologicalbiological-products-step-5_en.pdf (accessed on 13 January 2024).
- Harvey, R.D. Science of Biosimilars. J. Oncol. Pract. 2017, 13, 17s–23s. [Google Scholar] [CrossRef]
- Arandia, N.; Garate, J.I.; Mabe, J. Embedded Sensor Systems in Medical Devices: Requisites and Challenges Ahead. Sensors 2022, 22, 9917. [Google Scholar] [CrossRef]
- European Medicines Agency. From Laboratory to Patient: The Journey of a Centrally Authorised Medicine. EMA/103813/2018 Rev. 1. 2019. Available online: https://www.ema.europa.eu/en/documents/other/laboratory-patient-journey-centrally-authorised-medicine_en.pdf (accessed on 16 January 2024).
- What We Do. Available online: https://www.ema.europa.eu/en/about-us/what-we-do (accessed on 16 January 2024).
- Multidisciplinary: Biosimilar. Available online: https://www.ema.europa.eu/en/human-regulatory-overview/research-and-development/scientific-guidelines/multidisciplinary-guidelines/multidisciplinary-biosimilar (accessed on 3 August 2024).
- What We Do. Available online: https://www.fda.gov/about-fda/what-we-do (accessed on 17 January 2024).
- Biosimilars Guidances. Available online: https://www.fda.gov/vaccines-blood-biologics/general-biologics-guidances/biosimilars-guidances (accessed on 3 August 2024).
- Jordan, J.B.; Christl, L. FDA Biosimilar Action Plan: Could Improving Pharmacovigilance of Biologics Improve Patient and Physician Confidence in Biosimilars? Expert Opin. Drug Saf. 2020, 19, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, L.; Davies, M.; Home, P.; Forst, T.; Vilsbøll, T.; Schnell, O. Understanding Biosimilar Insulins—Development, Manufacturing, and Clinical Trials. J. Diabetes Sci. Technol. 2023, 17, 1649–1661. [Google Scholar] [CrossRef] [PubMed]
- About WHO. Available online: https://www.who.int/about (accessed on 30 May 2024).
- Mission. Available online: https://www.ich.org/page/mission (accessed on 30 May 2024).
- Niazi, S.K.; Al-Shaqha, W.M.; Mirza, Z. Proposal of International Council for Harmonization (ICH) Guideline for the Approval of Biosimilars. J. Mark. Access Health Policy 2023, 11, 2147286. [Google Scholar] [CrossRef] [PubMed]
- Radovan, D. Biosimilar development—An overview. Med. Writ. 2019, 28, 20–27. [Google Scholar]
- Vulto, A.G.; Jaquez, O.A. The Process Defines the Product: What Really Matters in Biosimilar Design and Production? Rheumatology 2017, 56, 14–29. [Google Scholar] [CrossRef]
- Amgen Biosimilars. Developing Biosimilars: The Process and Quality Standards. 2017. Available online: https://www.amgenoncology.com/resources/developing_biosimilars-USA-BIO-047538.pdf (accessed on 2 February 2024).
- Yu, L.X.; Amidon, G.; Khan, M.A.; Hoag, S.W.; Polli, J.; Raju, G.K.; Woodcock, J. Understanding Pharmaceutical Quality by Design. AAPS J. 2014, 16, 771–783. [Google Scholar] [CrossRef]
- Alsamil, A.M.; Giezen, T.J.; Egberts, T.C.; Leufkens, H.G.; Vulto, A.G.; Van der Plas, M.R.; Gardarsdottir, H. Reporting of Quality Attributes in Scientific Publications Presenting Biosimilarity Assessments of (Intended) Biosimilars: A Systematic Literature Review. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2020, 154, 105501. [Google Scholar] [CrossRef]
- Gonçalves, J.; Araújo, F.; Cutolo, M.; Fonseca, J.E. Biosimilar Monoclonal Antibodies: Preclinical and Clinical Development Aspects. Clin. Exp. Rheumatol. 2016, 34, 698–705. [Google Scholar]
- Al-Sabbagh, A.; Olech, E.; McClellan, J.E.; Kirchhoff, C.F. Development of Biosimilars. Semin. Arthritis Rheum. 2016, 45, S11–S18. [Google Scholar] [CrossRef]
- Daller, J. Biosimilars: A Consideration of the Regulations in the United States and European Union. Regul. Toxicol. Pharmacol. 2016, 76, 199–208. [Google Scholar] [CrossRef]
- Dranitsaris, G.; Amir, E.; Dorward, K. Biosimilars of Biological Drug Therapies: Regulatory, Clinical and Commercial Considerations. Drugs 2011, 71, 1527–1536. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.F.; Ma, J.; Winter, C.; Bayer, R. Recovery and purification process development for monoclonal antibody production. mAbs 2010, 2, 480–499. [Google Scholar] [CrossRef] [PubMed]
- Shire, S.J. Formulation and Manufacturability of Biologics. Curr. Opin. Biotechnol. 2009, 20, 708–714. [Google Scholar] [CrossRef]
- European Medicines Agency. Guideline on Comparability of Biotechnology-Derived Medicinal Products after a Change in the Manufacturing Process. Non-Clinical and Clinical Issues. EMEA/CHMP/BMWP/101695/2006. 2007. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-comparability-biotechnology-derived-medicinal-products-after-change-manufacturing-process-non-clinical-and-clinical-issues_en.pdf (accessed on 19 March 2024).
- Food and Drug Administration. Scientific Considerations in Demonstrating Biosimilarity to a Reference Product: Guidance for Industry. 2015. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/scientific-considerations-demonstrating-biosimilarity-reference-product (accessed on 25 March 2024).
- Li, E.C.; Abbas, R.; Jacobs, I.A.; Yin, D. Considerations in the Early Development of Biosimilar Products. Drug Discov. Today 2015, 20, 1–9. [Google Scholar] [CrossRef]
- Amgen Biosimilars. Biologic Comparability Testing versus Demonstration of Biosimilarity. 2018. Available online: https://www.amgenoncology.com/resources/biosimilars-biologic-comparability-testing-vs-demonstration-of-biosimilarity-USA-BIO-061158.pdf (accessed on 28 March 2024).
- Kaiser, P.K.; Schmitz-Valckenberg, M.S.; Holz, F.G. Anti–Vascular Endothelial Growth Factor Biosimilars in Ophthalmology. Retina 2022, 42, 2243–2250. [Google Scholar] [CrossRef]
- European Medicines Agency. Guideline on Similar Biological Medicinal Products Containing Biotechnology-Derived Proteins as Active Substance: Quality Issues (Revision 1). EMA/CHMP/BWP/247713/2012. 2014. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active-substance-quality-issues-revision-1_en.pdf (accessed on 2 April 2024).
- European Medicines Agency. Guideline on Similar Biological Medicinal Products Containing Biotechnology-Derived Proteins as Active Substance: Non-Clinical and Clinical Issues. EMEA/CHMP/BMWP/42832/2005 Rev1. 2014. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active-substance-non-clinical-and-clinical-issues-revision-1_en.pdf (accessed on 5 April 2024).
- Sharma, A.; Khante, S.; Mahadik, K.R.; Gaikwad, V.L. Regulatory Perspective of International Agencies for Development of Biosimilar Products (Monoclonal Antibodies): An Overview. Ther. Innov. Regul. Sci. 2020, 54, 965–977. [Google Scholar] [CrossRef]
- Webster, C.J.; George, K.L.; Woollett, G.R. Comparability of Biologics: Global Principles, Evidentiary Consistency and Unrealized Reliance. BioDrugs Clin. Immunother. Biopharm. Gene Ther. 2021, 35, 379–387. [Google Scholar] [CrossRef]
- McCamish, M.; Woollett, G. The Continuum of Comparability Extends to Biosimilarity: How Much Is Enough and What Clinical Data Are Necessary? Clin. Pharmacol. Ther. 2013, 93, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Socinski, M.A.; Curigliano, G.; Jacobs, I.; Gumbiner, B.; MacDonald, J.; Thomas, D. Clinical Considerations for the Development of Biosimilars in Oncology. mAbs 2015, 7, 286–293. [Google Scholar] [CrossRef]
- Markus, R.; Liu, J.; Ramchandani, M.; Landa, D.; Born, T.; Kaur, P. Developing the Totality of Evidence for Biosimilars: Regulatory Considerations and Building Confidence for the Healthcare Community. BioDrugs Clin. Immunother. Biopharm. Gene Ther. 2017, 31, 175–187. [Google Scholar] [CrossRef]
- Mascarenhas-Melo, F.; Diaz, M.; Gonçalves, M.B.S.; Vieira, P.; Bell, V.; Viana, S.; Nunes, S.; Paiva-Santos, A.C.; Veiga, F. An Overview of Biosimilars-Development, Quality, Regulatory Issues, and Management in Healthcare. Pharmaceuticals 2024, 17, 235. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S.; Linton, K.M.; Cervi, P.; Rosenberg, J.A.; Jacobs, I. A Clinician’s Guide to Biosimilars in Oncology. Cancer Treat. Rev. 2016, 46, 73–79. [Google Scholar] [CrossRef]
- Blank, T.; Vogt-Eisele, A.; Kaszkin-Bettag, M.; Netzer, T.; Hildebrandt, W. Safety and toxicity of biosimilars—EU versus US regulation. BaBI J. 2013, 2, 144–150. [Google Scholar] [CrossRef]
- Mellstedt, H.; Niederwieser, D.; Ludwig, H. The Challenge of Biosimilars. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2008, 19, 411–419. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, R.; Ward, M. Safety considerations of biosimilars. Aust. Prescr. 2016, 39, 188–189. [Google Scholar] [CrossRef]
- Kurki, P.; Barry, S.; Bourges, I.; Tsantili, P.; Wolff-Holz, E. Safety, Immunogenicity and Interchangeability of Biosimilar Monoclonal Antibodies and Fusion Proteins: A Regulatory Perspective. Drugs 2021, 81, 1881–1896. [Google Scholar] [CrossRef] [PubMed]
- Schreitmüller, T.; Barton, B.; Zharkov, A.; Bakalos, G. Comparative Immunogenicity Assessment of Biosimilars. Future Oncol. 2019, 15, 319–329. [Google Scholar] [CrossRef]
- European Medicines Agency. Guideline on Immunogenicity Assessment of Therapeutic Proteins. EMEA/CHMP/BMWP/14327/2006 Rev 1. 2017. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-immunogenicity-assessment-therapeutic-proteins-revision-1_en.pdf (accessed on 19 April 2024).
- Amgen Biosimilars. Determining Immunogenic Potential. 2018. Available online: https://www.amgenoncology.com/resources/biosimilars-determining-immunogenic-potential-USA-BIO-80007.pdf (accessed on 22 April 2024).
- Weise, M.; Bielsky, M.C.; Smet, K.D.; Ehmann, F.; Ekman, N.; Giezen, T.J.; Gravanis, I.; Heim, H.K.; Heinonen, E.; Ho, K.; et al. Biosimilars: What Clinicians Should Know. Blood 2012, 120, 5111–5117. [Google Scholar] [CrossRef]
- Weise, M.; Kurki, P.; Wolff-Holz, E.; Bielsky, M.C.; Schneider, C.K. Biosimilars: The Science of Extrapolation. Blood 2014, 124, 3191–3196. [Google Scholar] [CrossRef]
- Amgen Biosimilars. Extrapolation of Indications. 2018. Available online: https://www.amgenoncology.com/resources/biosimilars-extrapolation-of-indications-USA-BIO-061159.pdf (accessed on 27 April 2024).
- Curigliano, G.; O’Connor, D.P.; Rosenberg, J.A.; Jacobs, I. Biosimilars: Extrapolation for Oncology. Crit. Rev. Oncol./Hematol. 2016, 104, 131–137. [Google Scholar] [CrossRef]
- Kabir, E.R.; Moreino, S.S.; Siam, M.K.S. The Breakthrough of Biosimilars: A Twist in the Narrative of Biological Therapy. Biomolecules 2019, 9, 410. [Google Scholar] [CrossRef]
- Chao, J.; Skup, M.; Alexander, E.; Tundia, N.; Macaulay, D.; Wu, E.; Mulani, P. Nomenclature and Traceability Debate for Biosimilars: Small-Molecule Surrogates Lend Support for Distinguishable Nonproprietary Names. Adv. Ther. 2015, 32, 270–283. [Google Scholar] [CrossRef]
- Isaacs, J.; Goncalves, J.; Strohal, R.; Castañeda-Hernández, G.; Azevedo, V.; Dörner, T.; McInnes, I. The biosimilar approval process: How different is it? Consid. Med. 2017, 1, 3–6. [Google Scholar] [CrossRef]
- European Medicines Agency. Statement on the Scientific Rationale Supporting Interchangeability of Biosimilar Medicines in the EU. EMA/627319/2022. 2023. Available online: https://www.ema.europa.eu/en/documents/public-statement/statement-scientific-rationale-supporting-interchangeability-biosimilar-medicines-eu_en.pdf (accessed on 14 May 2024).
- Kurki, P.; Aerts, L.V.; Wolff-Holz, E.; Giezen, T.; Skibeli, V.; Weise, M. Interchangeability of Biosimilars: A European Perspective. BioDrugs Clin. Immunother. Biopharm. Gene Ther. 2017, 31, 83–91. [Google Scholar] [CrossRef]
- Cordeiro, M.A.; Vitorino, C.; Sinogas, C.; Sousa, J.J. A Regulatory Perspective on Biosimilar Medicines. Pharmaceutics 2024, 16, 321. [Google Scholar] [CrossRef]
- Oliveira, R.; Aires, T. Biossimilares: Velhas Questões, Novos Desafios. Gaz. Méd. 2016, 3. [Google Scholar] [CrossRef]
- Comissão Nacional de Farmácia e Terapêutica. Utilização de Medicamentos Biossimilares e Mudança de Medicamento Biológico de Referência para um Biossimilar. 2018. Available online: https://www.infarmed.pt/documents/15786/1816213/Orienta%20%C3%A7%C3%A3o+n+%C2%BA+5+-+Utiliza%C3%A7%C3%A3o+de+medicamentos%20+biossimilares/ddf4797c-a329-40d7-8d92-a1635a2f2ea7 (accessed on 21 May 2024).
- Food and Drug Administration. Biosimilar Regulatory Review and Approval. Available online: https://www.fda.gov/media/151061/download (accessed on 28 May 2024).
- Afzali, A.; Furtner, D.; Melsheimer, R.; Molloy, P.J. The Automatic Substitution of Biosimilars: Definitions of Interchangeability Are Not Interchangeable. Adv. Ther. 2021, 38, 2077–2093. [Google Scholar] [CrossRef]
- Albersmeyer, U.; Malessa, R.; Storz, U. Patenting Small and Large Pharmaceutical Molecules. Success. Drug Discov. 2018, 3, 41–64. [Google Scholar] [CrossRef]
- Raveendrashenoy, R.T. Role of Patents in Biosimilar Drug Development and Public Interest. J. Scientometr. Res. 2020, 9, s37–s47. [Google Scholar] [CrossRef]
- European Medicines Agency. ICH Guideline M4 (R4) on Common Technical Document (CTD) for the Registration of Pharmaceuticals for Human Use—Organisation of CTD. EMA/CPMP/ICH/2887/1999. 2021. Available online: https://www.ema.europa.eu/system/files/documents/scientific-guideline/m4_step_5_ctd_for_the_registration_of_pharmaceuticals_for_human_use_-_organisation_of_ctd-en.pdf (accessed on 3 June 2024).
- Garg, V.; Chopra, S.; Singh, S.K.; Gulati, M.; Kumar, B.; Mittal, N. A comparative study of common technical document in different regulated market. J. Pharm. Res. 2017, 11, 1015–1024. [Google Scholar]
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). ICH M8—Specification for Submission Formats for eCTD v1.3. 2021. Available online: https://admin.ich.org/sites/default/files/inline-files/Specification_for_Submission_Formats_for_eCTD_v1_3.pdf (accessed on 7 June 2024).
- Zuñiga, L.; Calvo, B. Biosimilars Approval Process. Regul. Toxicol. Pharmacol. 2010, 56, 374–377. [Google Scholar] [CrossRef] [PubMed]
- Bui, L.A.; Hurst, S.; Finch, G.L.; Ingram, B.; Jacobs, I.A.; Kirchhoff, C.F.; Ng, C.K.; Ryan, A.M. Key Considerations in the Preclinical Development of Biosimilars. Drug Discov. Today 2015, 20, 3–15. [Google Scholar] [CrossRef]
- Iqbal, Z.; Sadaf, S. Biosimilars: A Comparative Study of Regulatory, Safety and Pharmacovigilance Monograph in the Developed and Developing Economies. J. Pharm. Pharm. Sci. 2022, 25, 149–182. [Google Scholar] [CrossRef]
- Declerck, P.J. Biologicals and biosimilars: A review of the science and its implications. GaBI J. 2012, 1, 13–16. [Google Scholar] [CrossRef]
- Mysler, E.; Pineda, C.; Horiuchi, T.; Singh, E.; Mahgoub, E.; Coindreau, J.; Jacobs, I. Clinical and Regulatory Perspectives on Biosimilar Therapies and Intended Copies of Biologics in Rheumatology. Rheumatol. Int. 2016, 36, 613–625. [Google Scholar] [CrossRef]
- European Commission. Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community Code Relating to Medicinal Products for Human Use. Official Journal of the European Communities. 2001. Available online: https://eur-lex.europa.eu/legal-content/en/TXT/?uri=CELEX%3A32001L0083 (accessed on 12 June 2024).
- European Medicines Agency. The European Regulatory System for Medicines—Bringing New Safe and Effective Medicines to Patients across the European Union. 2023. Available online: https://www.ema.europa.eu/en/documents/leaflet/european-regulatory-system-medicines_en.pdf (accessed on 12 June 2024).
- European Commission. Regulation (EC) No 726/2004 of the European Parliament and of the Council of 31 March 2004 Laying Down Community Procedures for the Authorisation and Supervision of Medicinal Products for Human and Veterinary Use and Establishing a European Medicines Agency. Official Journal of the European Union. 2004. Available online: https://eur-lex.europa.eu/eli/reg/2004/726/oj (accessed on 12 June 2024).
- Authorisation of Medicines. Available online: https://www.ema.europa.eu/en/about-us/what-we-do/authorisation-medicines (accessed on 15 June 2024).
- Abed, I. The Approval Process of Medicines in Europe. Med. Writ. 2014, 23, 117–121. [Google Scholar] [CrossRef]
- European Commission. Commission Directive 2003/63/EC of 25 June 2003 Amending Directive 2001/83/EC of the European Parliament and of the Council on the Community Code Relating to Medicinal Products for Human Use. Official Journal of the European Union. 2003. Available online: https://eur-lex.europa.eu/legal-content/en/ALL/?uri=CELEX%3A32003L0063 (accessed on 15 June 2024).
- European Commission. Directive 2004/27/EC of the European Parliament and of the Council of 31 March 2004 Amending Directive 2001/83/EC on the Community Code Relating to Medicinal Products for Human Use. Official Journal of the European Union. 2004. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A32004L0027 (accessed on 15 June 2024).
- European Medicines Agency. European Medicines Agency Procedural Advice for Users of the Centralised Procedure for Similar Biological Medicinal Products Applications. EMA/940451/2011. 2019. Available online: https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/european-medicines-agency-procedural-advice-users-centralised-procedure-similar-biological-medicinal-product-applications_en.pdf (accessed on 21 June 2024).
- Vikram; Deep, A.; Manita. Regulation and Challenges of Biosimilars in European Union. Appl. Clin. Res. Clin. Trials Regul. Aff. 2019, 6, 192–211. [Google Scholar] [CrossRef]
- Pashikanti, S.; Vanacharla, J.S.D.; Sowmya, A.N.V.L. Regulatory overview of biosimilars in Europe. Int. J. Drug Regul. Aff. 2018, 6, 40–44. [Google Scholar] [CrossRef]
- Sheridan, M.; Massich, M.; Ashourian, N. Biosimilars: From Production to Patient. J. Infus. Nurs. Off. Publ. Infus. Nurses Soc. 2024, 47, 19–29. [Google Scholar] [CrossRef]
- Mohammed, Y.M. Regulatory pathways for development and submission activities. Med. Writ. 2019, 28, 8–19. [Google Scholar]
- Lemery, S.J.; Ricci, M.S.; Keegan, P.; McKee, A.E.; Pazdur, R. FDA’s Approach to Regulating Biosimilars. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 1882–1885. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. General Licensing Provisions; Section 351(k) Biosimilar Applications. Fed. Regist. 2018, 83, 58583–58585. [Google Scholar]
- Burchiel, S.W.; Aspbury, R.; Munday, J. The Search for Biosimilars and Biobetters. Drug Discov. Today 2019, 24, 1087–1091. [Google Scholar] [CrossRef]
- Food and Drug Administration. CDER 21st Century Review Process Desk Reference Guide. 2014. Available online: https://www.fda.gov/media/78941/download (accessed on 28 June 2024).
- Christl, L.A.; Woodcock, J.; Kozlowski, S. Biosimilars: The US Regulatory Framework. Annu. Rev. Med. 2017, 68, 243–254. [Google Scholar] [CrossRef]
- Purple Book-Database of Licensed Biological Products. Available online: https://purplebooksearch.fda.gov/ (accessed on 29 June 2024).
- Biosimilar Medicines. Available online: https://www.ema.europa.eu/en/search?search_api__fulltext=biosimilar&search_api_fulltext=biosimilar%20medicines (accessed on 3 August 2024).
- FDA-Approved Biosimilar Products. Available online: https://www.fda.gov/drugs/biosimilars/biosimilar-product-information (accessed on 3 August 2024).
- Metko, D.; Torres, T.; Vender, R. Viewpoint about biologic agents for psoriasis: Are they immunosuppressants or immunomodulators? J. Int. Med. Res. 2023, 51, 03000605231175547. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regulatory Authority | Biosimilar Medicine Definition |
|---|---|
| EMA | “Biological medicine highly similar to another biological medicine already approved in the EU” [2]. |
| FDA | “Biological product that is highly similar to and has no clinically meaningful differences from an existing FDA-approved reference product” [12]. |
| Topic | Title |
|---|---|
| Tailored clinical approach for biosimilars | Concept paper for the development of a reflection paper on a tailored clinical approach in biosimilar development. |
| Overarching biosimilar guidelines | Similar biological medicinal products. Similar biological medicinal products containing biotecnology-derived proteins as active substance: non-clinical and clinical issues. Similar biological medicinal products containing biotecnology-derived proteins as active substance: quality issues. |
| Product-specific biosimilar guidelines | Biosimilar medicinal products containing recombinant granulocyte-colony stimulating factor. Non-clinical and clinical development of similar biological medicinal products containing low-molecular-weight heparins. Non-clinical and clinical development of similar biological medicinal products containing recombinant human insulin and insulin analogues. Similar biological medicinal products containing interferon beta. Similar biological medicinal products containing monoclonal antibodies: non-clinical and clinical issues. Similar biological medicinal products containing recombinant erythropoietins. Similar biological medicinal products containing recombinant follicle-stimulating hormone. Similar medicinal products containing somatropin. Reflection paper: Non-clinical and clinical development of similar medicinal products containing recombinant Interferon Alfa. |
| Other guidelines relevant for biosimilars | Comparability of biotechnology-derived medicinal products after a change in the manufacturing process—non-clinical and clinical issues. Immunogenicity assessment of biotechnology-derived therapeutic proteins. Immunogenicity assessment of monoclonal antibodies intended for in vivo clinical use. Biosimilars—What are the key pharmacokinetic considerations in the assessment of biosimilarity? |
| Year | Title |
|---|---|
| 2014 | Reference Product Exclusivity for Biological Products Filed Under; Draft Guidance for Industry. |
| 2015 | Quality Considerations in Demonstrating Biosimilarity of a Therapeutic Protein Product to a Reference Product; Guidance for Industry. Scientific Considerations in Demonstrating Biosimilarity to a Reference Product; Guidance for Industry. |
| 2016 | Clinical Pharmacology Data to Support a Demonstration of Biosimilarity to a Reference Product; Guidance for Industry. |
| 2018 | Labelling for Biosimilar Products; Guidance for Industry. |
| 2019 | Considerations in Demonstrating Interchangeability with a Reference Product; Guidance for Industry. Development of Therapeutic Protein Biosimilars: Comparative Analytical Assessment and Other Quality-Related Considerations; Draft Guidance for Industry. |
| 2020 | Biosimilars and Interchangeable Biosimilars: Licensure for Fewer Than All Conditions of Use for Which the Reference Product Has Been Licensed; Draft Guidance for Industry. Biosimilarity and Interchangeability: Additional Draft Q&As on Biosimilar Development and the BPCI Act; Draft Guidance for Industry. |
| 2021 | New and Revised Draft Q&As on Biosimilar Development and the BPCI Act (Revision 3); Draft Guidance for Industry. Questions and Answers on Biosimilar Development and the BPCI Act; Guidance for Industry. |
| 2022 | Expansion Cohorts: Use in First-In-Human Clinical Trials to Expedite Development of Oncology Drugs and Biologics; Guidance for Industry. |
| 2023 | Classification Categories for Certain Supplements Under BsUFA III; Draft Guidance for Industry. Labeling for Biosimilar and Interchangeable Biosimilar Products; Draft Guidance for Industry. Biosimilarity and Interchangeability: Additional Draft Q&As on Biosimilar Development and the BPCI Act (Revision 1); Draft Guidance for Industry. |
| 2024 | Considerations in Demonstrating Interchangeability with a Reference Product: Update; Draft Guidance for Industry. Postapproval Manufacturing Changes to Biosimilar and Interchangeable Biosimilar Products Questions and Answers; Draft Guidance for Industry. |
| Attribute | Methods |
|---|---|
| Primary structure | Peptide mapping: liquid chromatography (LC) and mass spectrometry (MS). Peptide mass fingerprint: matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS). |
| Higher order structure | Nuclear magnetic resonance (NMR). Surface plasmon resonance (SPR). |
| Post-translational modifications | Normal-phase high-performance liquid chromatography coupled with mass spectrometry (NP-HPLC–MS). Gas chromatography–mass spectrometry (GC–MS). |
| Size, detection of aggregates | Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). High-performance size exclusion chromatography (HP-SEC). |
| Binding | Surface plasmon resonance (SPR). Enzyme-linked immunosorbent assay (ELISA). |
| Biological activity | Cell assays. In vivo assays. |
| Module 3 Quality | Module 4 Non-Clinical | Module 5 Clinical |
|---|---|---|
Drug Substance:
Drug Product:
| Pharmacology:
Pharmacokinetics:
Toxicology:
| Pharmacology Pharmacokinetics:
Pharmacodynamics:
Efficacy and safety:
Post-marketing:
|
| Mandatory Scope | Medicinal products developed by means of one of the following biotechnological processes:
|
| Optional Scope | Similar biological medicinal products of a centrally authorized product have automatic access to the CP. |
Similar biological medicinal products of a National/MRP/DCP product could, at the request of the applicant, be accepted for consideration under the CP, when the applicant shows that the medicinal product constitutes:
|
| EMA | FDA | |
|---|---|---|
| Approval Process | Centralized procedure | Section 351(k) of the PHSA |
| Scope | Valid at regional level, covering all 27 Member States of the EU and the countries of the EEA, Norway and Iceland. | Valid nationally, only within the US. |
| Teams Involved | BMWP, CHMP and PRAC. | CBER, CDER and OTBB. |
| Main Steps of the Evaluation | Pre-submission; assessment; Preparation of responses; Assessment of responses; Preparation of responses; Final CHMP opinion; Final EC decision. | Pre-submission activities; Process submission; Plan the review of the application; Conduct scientific/regulatory review; Official action on the application; Post-decision feedback. |
| Steps After the Evaluation | Transmission of Opinion and Annexes in all EU languages to Applicant, Commission, members of the Standing Committee, Norway and Iceland; EMA issues press releases and updates its official website to inform about the approval. | The final decision is communicated to all team members and to the applicant; The biosimilar is included in the Purple Book. |
| Time | 210 “active” days + periods when the clock is stopped. | About 10 months (~300 days). |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amaral, C.; Rodrigues, A.R.; Veiga, F.; Bell, V. Biosimilar Medicines: From Development Process to Marketing Authorization by the EMA and the FDA. Appl. Sci. 2024, 14, 7529. https://doi.org/10.3390/app14177529
Amaral C, Rodrigues AR, Veiga F, Bell V. Biosimilar Medicines: From Development Process to Marketing Authorization by the EMA and the FDA. Applied Sciences. 2024; 14(17):7529. https://doi.org/10.3390/app14177529
Chicago/Turabian StyleAmaral, Carolina, Ana Rita Rodrigues, Francisco Veiga, and Victoria Bell. 2024. "Biosimilar Medicines: From Development Process to Marketing Authorization by the EMA and the FDA" Applied Sciences 14, no. 17: 7529. https://doi.org/10.3390/app14177529