
Balance of Antioxidants vs. Oxidants in Perinatal Asphyxia
 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
The Literature Search Strategy
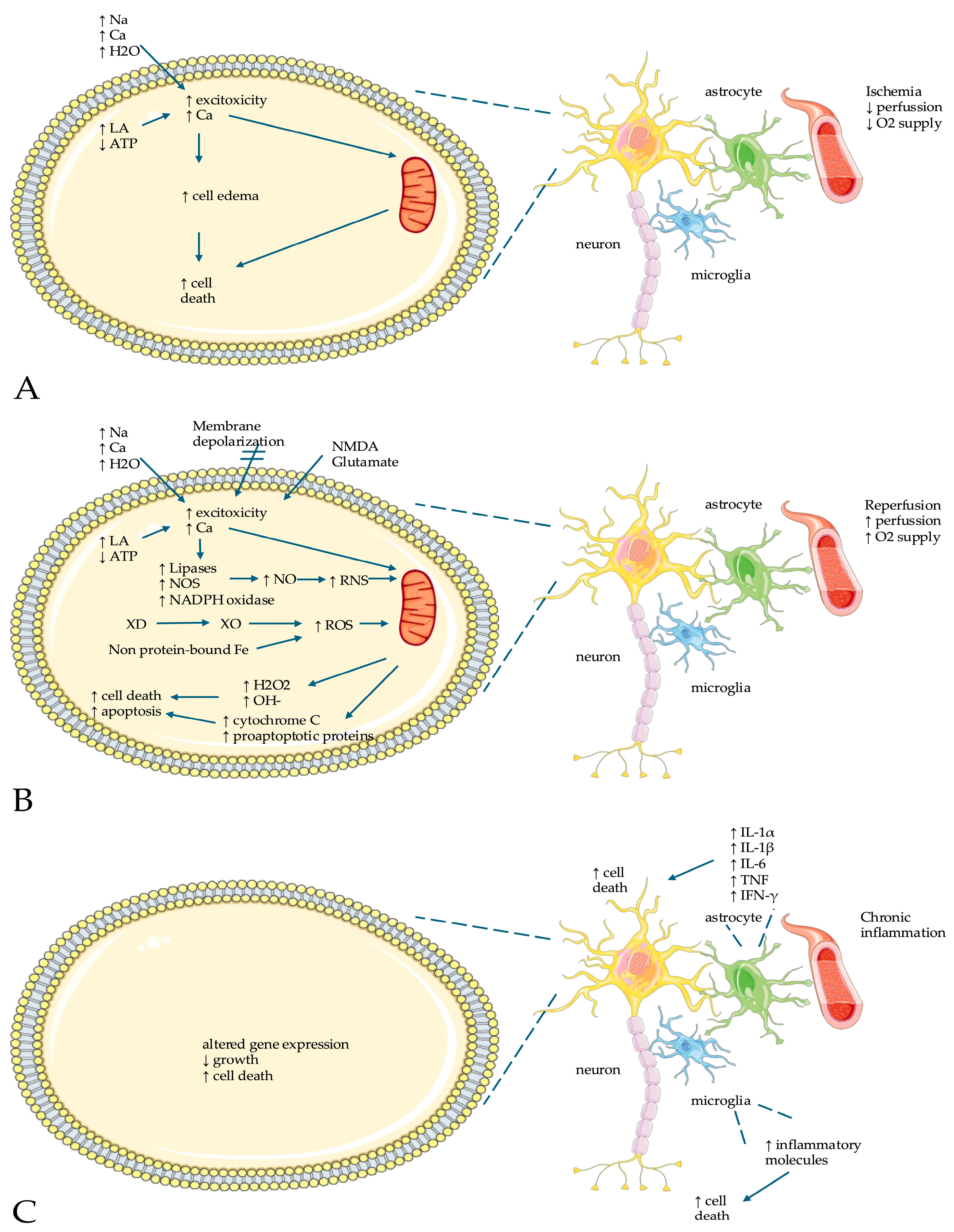
3. Pathophysiology
3.1. Aerobic Metabolism and Oxidative Phosphorylation
3.2. Biological Antioxidant Systems
3.3. Hypoxia/Ischemia Reperfusion Injury
4. Animal Models of Neonatal Hypoxia/Ischemia
5. Free Radical Biomarkers in NE
5.1. Enzymes Against Free Radicals
5.2. Nitric Oxide
5.3. Perfusion-Assisted Breathing
5.4. Markers of Lipid Damage (Malondialdehyde and 4-Hydroxynonenal)
5.5. Peroxidation-like Compounds
5.6. Protein Oxidation Markers
5.7. Uric Acid
5.8. Iron Not Bound by Proteins
6. Clinical Assessment of Neonates with Encephalopathy
7. Therapeutic Strategies
7.1. Hypothermia
7.2. Mitochondrial Therapy
7.3. Antioxidant Methods for Protecting the Brain from Injury Due to Low Oxygen Levels
7.4. Reduction in Free Radical Production Initiators and Scavengers
7.5. Nitric Oxide Inhibitors
7.6. Magnesium
7.7. Stem Cells
7.8. Nanomaterials
8. Discussion
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kurinczuk, J.J.; White-Koning, M.; Badawi, N. Epidemiology of neonatal encephalopathy and hypoxic-ischaemic encephalopathy. Early Hum. Dev. 2010, 86, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, G.; Pappas, A.; Shankaran, S. Outcomes in childhood following therapeutic hypothermia for neonatal hypoxic-ischemic encephalopathy (HIE). Semin. Perinatol. 2016, 40, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Marseglia, L.; D’Angelo, G.; Manti, S.; Arrigo, T.; Barberi, I.; Reiter, R.J.; Gitto, E. Oxidative Stress-Mediated Aging during the Fetal and Perinatal Periods. Oxidative Med. Cell. Longev. 2014, 2014, 358375. [Google Scholar] [CrossRef] [PubMed]
- Ozsurekci, Y.; Aykac, K.; Perrone, S. Oxidative Stress Related Diseases in Newborns. Oxidative Med. Cell. Longev. 2016, 2016, 2768365. [Google Scholar] [CrossRef]
- Berger, R.; Garnier, Y. Perinatal brain injury. J. Perinat. Med. 2000, 28, 261–285. [Google Scholar] [CrossRef]
- Qin, X.; Cheng, J.; Zhong, Y.; Mahgoub, O.K.; Akter, F.; Fan, Y.; Aldughaim, M.; Xie, Q.; Qin, L.; Gu, L.; et al. Mechanism and Treatment Related to Oxidative Stress in Neonatal Hypoxic-Ischemic Encephalopathy. Front. Mol. Neurosci. 2019, 12, 88. [Google Scholar] [CrossRef]
- Nuñez, A.; Benavente, I.; Blanco, D.; Boix, H.; Cabañas, F.; Chaffanel, M.; Fernández-Colomer, B.; Fernández-Lorenzo, J.R.; Loureiro, B.; Moral, M.T.; et al. Oxidative stress in perinatal asphyxia and hypoxic-ischaemic encephalopathy. An. Pediatría 2018, 88, 228.e1–228.e9. [Google Scholar] [CrossRef]
- Martini, S.; Austin, T.; Aceti, A.; Faldella, G.; Corvaglia, L. Free radicals and neonatal encephalopathy: Mechanisms of injury, biomarkers, and antioxidant treatment perspectives. Pediatr. Res. 2019, 87, 823–833. [Google Scholar] [CrossRef]
- Riljak, V.; Kraf, J.; Daryanani, A.; Jiruška, P.; Otáhal, J. Pathophysiology of Perinatal Hypoxic-Ischemic Encephalopathy—Biomarkers, Animal Models and Treatment Perspectives. Physiol. Res. 2016, 65, S533–S545. [Google Scholar] [CrossRef]
- Shi, Z.; Luo, K.; Deol, S.; Tan, S. A systematic review of noninflammatory cerebrospinal fluid biomarkers for clinical outcome in neonates with perinatal hypoxic brain injury that could be biologically significant. J. Neurosci. Res. 2021, 100, 2154–2173. [Google Scholar] [CrossRef]
- Kletkiewicz, H.; Klimiuk, M.; Woźniak, A.; Mila-Kierzenkowska, C.; Dokladny, K.; Rogalska, J. How to Improve the Antioxidant Defense in Asphyxiated Newborns—Lessons from Animal Models. Antioxidants 2020, 9, 898. [Google Scholar] [CrossRef] [PubMed]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, B. Teaching the basics of redox biology to medical and graduate students: Oxidants, antioxidants and disease mechanisms. Redox Biol. 2013, 1, 244–257. [Google Scholar] [CrossRef] [PubMed]
- Stamati, K.; Mudera, V.; Cheema, U. Evolution of oxygen utilization in multicellular organisms and implications for cell signalling in tissue engineering. J. Tissue Eng. 2011, 2, 2041731411432365. [Google Scholar] [CrossRef]
- Johnston, M.V.; Fatemi, A.; Wilson, M.A.; Northington, F. Treatment advances in neonatal neuroprotection and neurointensive care. Lancet Neurol. 2011, 10, 372–382. [Google Scholar] [CrossRef]
- Wassink, G.; Gunn, E.R.; Drury, P.P.; Bennet, L.; Gunn, A.J. The mechanisms and treatment of asphyxial encephalopathy. Front. Neurosci. 2014, 8, 40. [Google Scholar] [CrossRef]
- Torres-Cuevas, I.; Parra-Llorca, A.; Sánchez-Illana, A.; Nuñez-Ramiro, A.; Kuligowski, J.; Cháfer-Pericás, C.; Cernada, M.; Escobar, J.; Vento, M. Oxygen and oxidative stress in the perinatal period. Redox Biol. 2017, 12, 674–681. [Google Scholar] [CrossRef]
- Sonowal, R.; Jain, A.; Bhargava, V.; Khanna, H.D.; Kumar, A. Antioxidant Levels in Cord Blood of Term Low Birth Weight Neonates Requiring Delivery Room Resuscitation. J. Neonatol. 2021, 35, 20–23. [Google Scholar] [CrossRef]
- Spears, K.; Cheney, C.; Zerzan, J. Low plasma retinol concentrations increase the risk of developing bronchopulmonary dysplasia and long-term respiratory disability in very-low-birth-weight infants. Am. J. Clin. Nutr. 2004, 80, 1589–1594. [Google Scholar] [CrossRef]
- Jomova, K.; Raptova, R.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Reactive oxygen species, toxicity, oxidative stress, and antioxidants: Chronic diseases and aging. Arch. Toxicol. 2023, 97, 2499–2574. [Google Scholar] [CrossRef]
- Mutlu, M.; Sariaydin, M.; Aslan, Y.; Kader, S.; Dereci, S.; Kart, C.; Yaman, S.O.; Kural, B. Status of vitamin D, antioxidant enzymes, and antioxidant substances in neonates with neonatal hypoxic-ischemic encephalopathy. J. Matern. Fetal Neonatal Med. 2016, 29, 2259–2263. [Google Scholar] [CrossRef] [PubMed]
- Stessman, L.E.; Peeples, E.S. Vitamin D and Its Role in Neonatal Hypoxic-Ischemic Brain Injury. Neonatology 2018, 113, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Ibi, M.; Sawada, H.; Nakanishi, M.; Kume, T.; Katsuki, H.; Kaneko, S.; Shimohama, S.; Akaike, A. Protective effects of 1α,25-(OH)2D3 against the neurotoxicity of glutamate and reactive oxygen species in mesencephalic culture. Neuropharmacology 2001, 40, 761–771. [Google Scholar] [CrossRef]
- Marriott, L.D.; Foote, K.D.; Kimber, A.C.; Delves, H.T.; Morgan, J.B. Zinc, copper, selenium and manganese blood levels in preterm infants. Arch. Dis. Child. Fetal Neonatal Ed. 2007, 92, F494–F497. [Google Scholar] [CrossRef]
- Nassi, N.; Ponziani, V.; Becatti, M.; Galvan, P.; Donzelli, G. Anti-oxidant enzymes and related elements in term and preterm newborns. Pediatr. Int. 2009, 51, 183–187. [Google Scholar] [CrossRef]
- Altun, D.; Kurekci, A.E.; Gursel, O.; Hacıhamdioglu, D.O.; Kurt, I.; Aydın, A.; Ozcan, O. Malondialdehyde, Antioxidant Enzymes, and Renal Tubular Functions in Children with Iron Deficiency or Iron-Deficiency Anemia. Biol. Trace Elem. Res. 2014, 161, 48–56. [Google Scholar] [CrossRef]
- Boskabadi, H.; Navaee Boroujeni, A.; Mostafavi-Toroghi, H.; Hosseini, G.; Ghayour-Mobarhan, M.; Hamidi Alamdari, D.; Biranvandi, M.; Saber, H.; Ferns, G.A. Prooxidant-antioxidant balance in perinatal asphyxia. Indian J. Pediatr. 2014, 81, 248–253. [Google Scholar] [CrossRef]
- Leite, H.P.; Nogueira, P.C.K.; de Oliveira Iglesias, S.B.; de Oliveira, S.V.; Sarni, R.O.S. Increased plasma selenium is associated with better outcomes in children with systemic inflammation. Nutrition 2015, 31, 485–490. [Google Scholar] [CrossRef]
- Freitas, R.G.B.O.N.; Nogueira, R.J.N.; Antonio, M.A.R.G.M.; Barros-Filho, A.d.A.; Hessel, G. Selenium deficiency and the effects of supplementation on preterm infants. Rev. Paul. Pediatr. 2014, 32, 126–135. [Google Scholar] [CrossRef]
- Bizerea, T.; Dezsi, S.; Marginean, O.; Stroescu, R.; Rogobete, A.; Bizerea-Spiridon, O.; Ilie, C. The Link Between Selenium, Oxidative Stress and Pregnancy Induced Hypertensive Disorders. Clin. Lab. 2018, 64, 1593–1610. [Google Scholar] [CrossRef]
- Tara, F.; Rayman, M.P.; Boskabadi, H.; Ghayour-Mobarhan, M.; Sahebkar, A.; Alamdari, D.H.; Razavi, B.S.; Tavallaie, S.; Azimi-Nezhad, M.; Shakeri, M.T.; et al. Prooxidant-antioxidant balance in pregnancy: A randomized double-blind placebo-controlled trial of selenium supplementation. J. Perinat. Med. 2010, 38, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Tindell, R.; Tipple, T. Selenium: Implications for outcomes in extremely preterm infants. J. Perinatol. 2018, 38, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-D.; Zheng, W. Human Exposure and Health Effects of Inorganic and Elemental Mercury. J. Prev. Med. Public Health 2012, 45, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Boskabadi, H.; Ghayour-Mobarhan, M.; Saeidinia, A. Serum pro-oxidant/antioxidant balance in term versus preterm neonates. Medicine 2022, 101, e31381. [Google Scholar] [CrossRef]
- Keihanian, F.; Basirjafari, S.; Darbandi, B.; Saeidinia, A.; Jafroodi, M.; Sharafi, R.; Shakiba, M. Comparison of quantitative and qualitative tests for glucose-6-phosphate dehydrogenase deficiency in the neonatal period. Int. J. Lab. Hematol. 2017, 39, 251–260. [Google Scholar] [CrossRef]
- Ikonomidou, C.; Kaindl, A.M. Neuronal death and oxidative stress in the developing brain. Antioxid. Redox Signal 2011, 14, 1535–1550. [Google Scholar] [CrossRef]
- Rennie, J.M.; Hagmann, C.F.; Robertson, N.J. Outcome after intrapartum hypoxic ischaemia at term. Semin. Fetal Neonatal Med. 2007, 12, 398–407. [Google Scholar] [CrossRef]
- Millar, L.J.; Shi, L.; Hoerder-Suabedissen, A.; Molnar, Z. Neonatal Hypoxia Ischaemia: Mechanisms, Models, and Therapeutic Challenges. Front. Cell Neurosci. 2017, 11, 78. [Google Scholar] [CrossRef]
- Gunn, A.J.; Gunn, T.R.; de Haan, H.H.; Williams, C.E.; Gluckman, P.D. Dramatic neuronal rescue with prolonged selective head cooling after ischemia in fetal lambs. J. Clin. Investig. 1997, 99, 248–256. [Google Scholar] [CrossRef]
- Cotten, C.M.; Shankaran, S. Hypothermia for hypoxic-ischemic encephalopathy. Expert. Rev. Obstet. Gynecol. 2010, 5, 227–239. [Google Scholar] [CrossRef]
- Hope, P.L.; Cady, E.B.; Chu, A.; Delpy, D.T.; Gardiner, R.M.; Reynolds, E.O.R. Brain Metabolism and Intracellular pH During Ischaemia and Hypoxia: An In Vivo 31P and 1H Nuclear Magnetic Resonance Study in the Lamb. J. Neurochem. 2006, 49, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Zhu, P.; Fujino, M.; Zhuang, J.; Guo, H.; Sheikh, I.; Zhao, L.; Li, X.-K. Oxidative Stress in Hypoxic-Ischemic Encephalopathy: Molecular Mechanisms and Therapeutic Strategies. Int. J. Mol. Sci. 2016, 17, 2078. [Google Scholar] [CrossRef] [PubMed]
- Mayurasakorn, K.; Niatsetskaya, Z.V.; Sosunov, S.A.; Williams, J.J.; Zirpoli, H.; Vlasakov, I.; Deckelbaum, R.J.; Ten, V.S. DHA but Not EPA Emulsions Preserve Neurological and Mitochondrial Function after Brain Hypoxia-Ischemia in Neonatal Mice. PLoS ONE 2016, 11, e0160870. [Google Scholar] [CrossRef] [PubMed]
- Weidinger, A.; Kozlov, A. Biological Activities of Reactive Oxygen and Nitrogen Species: Oxidative Stress versus Signal Transduction. Biomolecules 2015, 5, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Signorini, C.; Perrone, S.; Sgherri, C.; Ciccoli, L.; Buonocore, G.; Leoncini, S.; Rossi, V.; Vecchio, D.; Comporti, M. Plasma esterified F2-isoprostanes and oxidative stress in newborns: Role of nonprotein-bound iron. Pediatr. Res. 2008, 63, 287–291. [Google Scholar] [CrossRef]
- McCord, J.M. Oxygen-derived free radicals in postischemic tissue injury. N. Engl. J. Med. 1985, 312, 159–163. [Google Scholar]
- Petrosillo, G.; Ruggiero, F.M.; Paradies, G. Role of reactive oxygen species and cardiolipin in the release of cytochrome c from mitochondria. FASEB J. 2003, 17, 2202–2208. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Nakka, V.P.; Gusain, A.; Mehta, S.L.; Raghubir, R. Molecular Mechanisms of Apoptosis in Cerebral Ischemia: Multiple Neuroprotective Opportunities. Mol. Neurobiol. 2007, 37, 7–38. [Google Scholar] [CrossRef]
- Douglas-Escobar, M.; Weiss, M.D. Hypoxic-ischemic encephalopathy: A review for the clinician. JAMA Pediatr. 2015, 169, 397–403. [Google Scholar] [CrossRef]
- Gunn, A.J.; Bennet, L. Timing still key to treating hypoxic ischaemic brain injury. Lancet Neurol. 2016, 15, 126–127. [Google Scholar] [CrossRef] [PubMed]
- Kaur, C.; Rathnasamy, G.; Ling, E.-A. Roles of Activated Microglia in Hypoxia Induced Neuroinflammation in the Developing Brain and the Retina. J. Neuroimmune Pharmacol. 2012, 8, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Jellema, R.K.; Lima Passos, V.; Ophelders, D.R.M.G.; Wolfs, T.G.A.M.; Zwanenburg, A.; De Munter, S.; Nikiforou, M.; Collins, J.J.P.; Kuypers, E.; Bos, G.M.J.; et al. Systemic G-CSF attenuates cerebral inflammation and hypomyelination but does not reduce seizure burden in preterm sheep exposed to global hypoxia–ischemia. Exp. Neurol. 2013, 250, 293–303. [Google Scholar] [CrossRef]
- Rice, J.E., 3rd; Vannucci, R.C.; Brierley, J.B. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann. Neurol. 1981, 9, 131–141. [Google Scholar] [CrossRef]
- Alexander, M.; Garbus, H.; Smith, A.L.; Rosenkrantz, T.S.; Fitch, R.H. Behavioral and histological outcomes following neonatal HI injury in a preterm (P3) and term (P7) rodent model. Behav. Brain Res. 2014, 259, 85–96. [Google Scholar] [CrossRef]
- Chen, W.; Jadhav, V.; Tang, J.; Zhang, J.H. HIF-1alpha inhibition ameliorates neonatal brain injury in a rat pup hypoxic-ischemic model. Neurobiol. Dis. 2008, 31, 433–441. [Google Scholar] [CrossRef]
- Vannucci, R.C.; Vannucci, S.J. Perinatal hypoxic-ischemic brain damage: Evolution of an animal model. Dev. Neurosci. 2005, 27, 81–86. [Google Scholar] [CrossRef]
- Jantzie, L.L.; Robinson, S. Preclinical Models of Encephalopathy of Prematurity. Dev. Neurosci. 2015, 37, 277–288. [Google Scholar] [CrossRef]
- Kyng, K.J.; Skajaa, T.; Kerrn-Jespersen, S.; Andreassen, C.S.; Bennedsgaard, K.; Henriksen, T.B. A Piglet Model of Neonatal Hypoxic-Ischemic Encephalopathy. J. Vis. Exp. 2015, 99, e52454. [Google Scholar] [CrossRef]
- Guerguerian, A.M.; Brambrink, A.M.; Traystman, R.J.; Huganir, R.L.; Martin, L.J. Altered expression and phosphorylation of N-methyl-D-aspartate receptors in piglet striatum after hypoxia-ischemia. Brain Res. Mol. Brain Res. 2002, 104, 66–80. [Google Scholar] [CrossRef]
- Broad, K.D.; Fierens, I.; Fleiss, B.; Rocha-Ferreira, E.; Ezzati, M.; Hassell, J.; Alonso-Alconada, D.; Bainbridge, A.; Kawano, G.; Ma, D.; et al. Inhaled 45-50% argon augments hypothermic brain protection in a piglet model of perinatal asphyxia. Neurobiol. Dis. 2016, 87, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Rogalska, J.; Danielisova, V.; Caputa, M. Effect of neonatal body temperature on postanoxic, potentially neurotoxic iron accumulation in the rat brain. Neurosci. Lett. 2006, 393, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.J.; Carini, M.; Butterfield, D.A. Protein carbonylation. Antioxid. Redox Signal 2010, 12, 323–325. [Google Scholar] [CrossRef] [PubMed]
- Barata, L.; Arruza, L.; Rodriguez, M.J.; Aleo, E.; Vierge, E.; Criado, E.; Sobrino, E.; Vargas, C.; Ceprian, M.; Gutierrez-Rodriguez, A.; et al. Neuroprotection by cannabidiol and hypothermia in a piglet model of newborn hypoxic-ischemic brain damage. Neuropharmacology 2019, 146, 1–11. [Google Scholar] [CrossRef]
- Santos, P.T.; O’Brien, C.E.; Chen, M.W.; Hopkins, C.D.; Adams, S.; Kulikowicz, E.; Singh, R.; Koehler, R.C.; Martin, L.J.; Lee, J.K. Proteasome Biology Is Compromised in White Matter After Asphyxic Cardiac Arrest in Neonatal Piglets. J. Am. Heart Assoc. 2018, 7, e009415. [Google Scholar] [CrossRef]
- Kletkiewicz, H.; Nowakowska, A.; Siejka, A.; Mila-Kierzenkowska, C.; Wozniak, A.; Caputa, M.; Rogalska, J. Deferoxamine prevents cerebral glutathione and vitamin E depletions in asphyxiated neonatal rats: Role of body temperature. Int. J. Hyperth. 2016, 32, 211–220. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, J.J.; Mei, Y.W.; Sun, S.G.; Tong, E.T. Effects of immediate and delayed mild hypothermia on endogenous antioxidant enzymes and energy metabolites following global cerebral ischemia. Chin. Med. J. 2011, 124, 2764–2766. [Google Scholar]
- Toader, A.M.; Filip, A.; Decea, N.; Muresan, A. Neuroprotective strategy in an experimental newborn rat model of brain ischemia and hypoxia: Effects of Resveratrol and hypothermia. Clujul Med. 2013, 86, 203–207. [Google Scholar]
- Chevin, M.; Guiraut, C.; Sebire, G. Effect of hypothermia on interleukin-1 receptor antagonist pharmacodynamics in inflammatory-sensitized hypoxic-ischemic encephalopathy of term newborns. J. Neuroinflammation 2018, 15, 214. [Google Scholar] [CrossRef]
- Dalen, M.L.; Alme, T.N.; Bjoras, M.; Munkeby, B.H.; Rootwelt, T.; Saugstad, O.D. Reduced expression of DNA glycosylases in post-hypoxic newborn pigs undergoing therapeutic hypothermia. Brain Res. 2010, 1363, 198–205. [Google Scholar] [CrossRef]
- Tan, S. Fault and blame, insults to the perinatal brain may be remote from time of birth. Clin. Perinatol. 2014, 41, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Derrick, M.; Englof, I.; Drobyshevsky, A.; Luo, K.; Yu, L.; Tan, S. Intrauterine fetal demise can be remote from the inciting insult in an animal model of hypoxia-ischemia. Pediatr. Res. 2012, 72, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Thornton, C.; Rousset, C.I.; Kichev, A.; Miyakuni, Y.; Vontell, R.; Baburamani, A.A.; Fleiss, B.; Gressens, P.; Hagberg, H. Molecular mechanisms of neonatal brain injury. Neurol. Res. Int. 2012, 2012, 506320. [Google Scholar] [CrossRef]
- Michel, T.M.; Camara, S.; Tatschner, T.; Frangou, S.; Sheldrick, A.J.; Riederer, P.; Grunblatt, E. Increased xanthine oxidase in the thalamus and putamen in depression. World J. Biol. Psychiatry 2010, 11, 314–320. [Google Scholar] [CrossRef]
- Tan, S.; Radi, R.; Gaudier, F.; Evans, R.A.; Rivera, A.; Kirk, K.A.; Parks, D.A. Physiologic levels of uric acid inhibit xanthine oxidase in human plasma. Pediatr. Res. 1993, 34, 303–307. [Google Scholar] [CrossRef]
- Kumar, A.; Ramakrishna, S.V.; Basu, S.; Rao, G.R. Oxidative stress in perinatal asphyxia. Pediatr. Neurol. 2008, 38, 181–185. [Google Scholar] [CrossRef]
- Gulcan, H.; Ozturk, I.C.; Arslan, S. Alterations in antioxidant enzyme activities in cerebrospinal fluid related with severity of hypoxic ischemic encephalopathy in newborns. Biol. Neonate 2005, 88, 87–91. [Google Scholar] [CrossRef]
- Gunes, T.; Ozturk, M.A.; Koklu, E.; Kose, K.; Gunes, I. Effect of allopurinol supplementation on nitric oxide levels in asphyxiated newborns. Pediatr. Neurol. 2007, 36, 17–24. [Google Scholar] [CrossRef]
- Kumar, A.; Mittal, R.; Khanna, H.D.; Basu, S. Free radical injury and blood-brain barrier permeability in hypoxic-ischemic encephalopathy. Pediatrics 2008, 122, e722–e727. [Google Scholar] [CrossRef]
- Boskabadi, H.; Moeini, M.; Tara, F.; Tavallaie, S.; Saber, H.; Nejati, R.; Hosseini, G.; Mostafavi-Toroghi, H.; Ferns, G.A.A.; Ghayour-Mobarhan, M. Determination of Prooxidant–Antioxidant Balance during Uncomplicated Pregnancy Using a Rapid Assay. J. Med. Biochem. 2013, 32, 227–232. [Google Scholar] [CrossRef]
- Ghayour-Mobarhan, M.; Alamdari, D.H.; Moohebati, M.; Sahebkar, A.; Nematy, M.; Safarian, M.; Azimi-Nezhad, M.; Reza Parizadeh, S.M.; Tavallaie, S.; Koliakos, G.; et al. Determination of Prooxidant—Antioxidant Balance After Acute Coronary Syndrome Using a Rapid Assay: A Pilot Study. Angiology 2009, 60, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Parizadeh, M.R.; Azarpazhooh, M.R.; Mobarra, N.; Nematy, M.; Alamdari, D.H.; Tavalaie, S.; Sahebkar, A.; Hassankhani, B.; Ferns, G.; Ghayour-Mobarhan, M. Prooxidant-antioxidant balance in stroke patients and 6-month prognosis. Clin. Lab. 2011, 57, 183–191. [Google Scholar]
- Boskabadi, H.; Zakerihamidi, M.; Heidarzadeh, M.; Avan, A.; Ghayour-Mobarhan, M.; Ferns, G.A. The value of serum pro-oxidant/antioxidant balance in the assessment of asphyxia in term neonates. J. Matern. Fetal Neonatal Med. 2017, 30, 1556–1561. [Google Scholar] [CrossRef]
- Banupriya, C.; Ratnakar; Doureradjou, P.; Mondal, N.; Vishnu, B.; Koner, B.C. Can urinary excretion rate of malondialdehyde, uric acid and protein predict the severity and impending death in perinatal asphyxia? Clin. Biochem. 2008, 41, 968–973. [Google Scholar] [CrossRef]
- Mondal, N.; Bhat, B.V.; Banupriya, C.; Koner, B.C. Oxidative stress in perinatal asphyxia in relation to outcome. Indian. J. Pediatr. 2010, 77, 515–517. [Google Scholar] [CrossRef]
- Shouman, B.O.; Mesbah, A.; Aly, H. Iron metabolism and lipid peroxidation products in infants with hypoxic ischemic encephalopathy. J. Perinatol. 2008, 28, 487–491. [Google Scholar] [CrossRef]
- Schmidt, H.; Grune, T.; Muller, R.; Siems, W.G.; Wauer, R.R. Increased levels of lipid peroxidation products malondialdehyde and 4-hydroxynonenal after perinatal hypoxia. Pediatr. Res. 1996, 40, 15–20. [Google Scholar] [CrossRef]
- Zelzer, S.; Mangge, H.; Oberreither, R.; Bernecker, C.; Gruber, H.J.; Pruller, F.; Fauler, G. Oxidative stress: Determination of 4-hydroxy-2-nonenal by gas chromatography/mass spectrometry in human and rat plasma. Free Radic. Res. 2015, 49, 1233–1238. [Google Scholar] [CrossRef]
- Sakamoto, H.; Corcoran, T.B.; Laffey, J.G.; Shorten, G.D. Isoprostanes--markers of ischaemia reperfusion injury. Eur. J. Anaesthesiol. 2002, 19, 550–559. [Google Scholar]
- Chafer-Pericas, C.; Cernada, M.; Rahkonen, L.; Stefanovic, V.; Andersson, S.; Vento, M. Preliminary case control study to establish the correlation between novel peroxidation biomarkers in cord serum and the severity of hypoxic ischemic encephalopathy. Free Radic. Biol. Med. 2016, 97, 244–249. [Google Scholar] [CrossRef]
- Negro, S.; Benders, M.; Tataranno, M.L.; Coviello, C.; de Vries, L.S.; van Bel, F.; Groenendaal, F.; Longini, M.; Proietti, F.; Belvisi, E.; et al. Early Prediction of Hypoxic-Ischemic Brain Injury by a New Panel of Biomarkers in a Population of Term Newborns. Oxid. Med. Cell Longev. 2018, 2018, 7608108. [Google Scholar] [CrossRef] [PubMed]
- Buonocore, G.; Perrone, S.; Longini, M.; Terzuoli, L.; Bracci, R. Total hydroperoxide and advanced oxidation protein products in preterm hypoxic babies. Pediatr. Res. 2000, 47, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.P.; Makadia, M.G.; Patel, V.I.; Nilayangode, H.N.; Nimbalkar, S.M. Urinary Uric Acid/Creatinine Ratio—A Marker For Perinatal Asphyxia. J. Clin. Diagn. Res. 2017, 11, SC08–SC10. [Google Scholar] [CrossRef]
- Dorrepaal, C.A.; Berger, H.M.; Benders, M.J.; van Zoeren-Grobben, D.; Van de Bor, M.; Van Bel, F. Nonprotein-bound iron in postasphyxial reperfusion injury of the newborn. Pediatrics 1996, 98, 883–889. [Google Scholar] [CrossRef]
- Lally, P.J.; Montaldo, P.; Oliveira, V.; Soe, A.; Swamy, R.; Bassett, P.; Mendoza, J.; Atreja, G.; Kariholu, U.; Pattnayak, S.; et al. Magnetic resonance spectroscopy assessment of brain injury after moderate hypothermia in neonatal encephalopathy: A prospective multicentre cohort study. Lancet Neurol. 2019, 18, 35–45. [Google Scholar] [CrossRef]
- Gunn, A.J.; Wyatt, J.S.; Whitelaw, A.; Barks, J.; Azzopardi, D.; Ballard, R.; Edwards, A.D.; Ferriero, D.M.; Gluckman, P.D.; Polin, R.A.; et al. Therapeutic Hypothermia Changes the Prognostic Value of Clinical Evaluation of Neonatal Encephalopathy. J. Pediatr. 2008, 152, 55–58.e51. [Google Scholar] [CrossRef]
- Mendler, M.R.; Mendler, I.; Hassan, M.A.; Mayer, B.; Bode, H.; Hummler, H.D. Predictive Value of Thompson-Score for Long-Term Neurological and Cognitive Outcome in Term Newborns with Perinatal Asphyxia and Hypoxic-Ischemic Encephalopathy Undergoing Controlled Hypothermia Treatment. Neonatology 2018, 114, 341–347. [Google Scholar] [CrossRef]
- Malin, G.L.; Morris, R.K.; Khan, K.S. Strength of association between umbilical cord pH and perinatal and long term outcomes: Systematic review and meta-analysis. BMJ 2010, 340, c1471. [Google Scholar] [CrossRef]
- Wayock, C.P.; Meserole, R.L.; Saria, S.; Jennings, J.M.; Huisman, T.A.G.M.; Northington, F.J.; Graham, E.M. Perinatal risk factors for severe injury in neonates treated with whole-body hypothermia for encephalopathy. Am. J. Obstet. Gynecol. 2014, 211, 41.e1–41.e8. [Google Scholar] [CrossRef]
- Pappas, A.; Shankaran, S.; Laptook, A.R.; Langer, J.C.; Bara, R.; Ehrenkranz, R.A.; Goldberg, R.N.; Das, A.; Higgins, R.D.; Tyson, J.E.; et al. Hypocarbia and Adverse Outcome in Neonatal Hypoxic-Ischemic Encephalopathy. J. Pediatr. 2011, 158, 752–758.e751. [Google Scholar] [CrossRef]
- Hansen, G.; Al Shafouri, N.; Narvey, M.; Vallance, J.K.; Srinivasan, G. High blood carbon dioxide variability and adverse outcomes in neonatal hypoxic ischemic encephalopathy. J. Matern. Fetal Neonatal Med. 2015, 29, 680–683. [Google Scholar] [CrossRef] [PubMed]
- Murray, D.M.; Boylan, G.B.; Fitzgerald, A.P.; Ryan, C.A.; Murphy, B.P.; Connolly, S. Persistent lactic acidosis in neonatal hypoxic-ischaemic encephalopathy correlates with EEG grade and electrographic seizure burden. Arch. Dis. Child. Fetal Neonatal Ed. 2008, 93, F183–F186. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.K.; Kaiser, J.R.; Guffey, D.; Minard, C.G.; Guillet, R.; Gunn, A.J. Hypoglycaemia and hyperglycaemia are associated with unfavourable outcome in infants with hypoxic ischaemic encephalopathy: A post hoc analysis of the CoolCap Study. Arch. Dis. Child. Fetal Neonatal Ed. 2016, 101, F149–F155. [Google Scholar] [CrossRef] [PubMed]
- Thoresen, M.; Hellström-Westas, L.; Liu, X.; de Vries, L.S. Effect of Hypothermia on Amplitude-Integrated Electroencephalogram in Infants With Asphyxia. Pediatrics 2010, 126, e131–e139. [Google Scholar] [CrossRef]
- Azzopardi, D. Clinical applications of cerebral function monitoring in neonates. Semin. Fetal Neonatal Med. 2015, 20, 154–163. [Google Scholar] [CrossRef]
- De Wispelaere, L.A.T.T.; Ouwehand, S.; Olsthoorn, M.; Govaert, P.; Smit, L.S.; de Jonge, R.C.J.; Lequin, M.H.; Reiss, I.K.; Dudink, J. Electroencephalography and brain magnetic resonance imaging in asphyxia comparing cooled and non-cooled infants. Eur. J. Paediatr. Neurol. 2019, 23, 181–190. [Google Scholar] [CrossRef]
- Taylor, M.J.; Murphy, W.J.; Whyte, H.E. Prognostic Reliability of Somatosensory and Visual Evoked Potentials of Asphyxiated Term Infants. Dev. Med. Child Neurol. 2008, 34, 507–515. [Google Scholar] [CrossRef]
- Nakamura, S.; Koyano, K.; Jinnai, W.; Hamano, S.; Yasuda, S.; Konishi, Y.; Kuboi, T.; Kanenishi, K.; Nishida, T.; Kusaka, T. Simultaneous measurement of cerebral hemoglobin oxygen saturation and blood volume in asphyxiated neonates by near-infrared time-resolved spectroscopy. Brain Dev. 2015, 37, 925–932. [Google Scholar] [CrossRef]
- Szakmar, E.; Smith, J.; Yang, E.; Volpe, J.J.; Inder, T.; El-Dib, M. Association between cerebral oxygen saturation and brain injury in neonates receiving therapeutic hypothermia for neonatal encephalopathy. J. Perinatol. 2021, 41, 269–277. [Google Scholar] [CrossRef]
- Thayyil, S.; Chandrasekaran, M.; Taylor, A.; Bainbridge, A.; Cady, E.B.; Chong, W.K.K.; Murad, S.; Omar, R.Z.; Robertson, N.J. Cerebral Magnetic Resonance Biomarkers in Neonatal Encephalopathy: A Meta-analysis. Pediatrics 2010, 125, e382–e395. [Google Scholar] [CrossRef]
- Rollins, N.; Booth, T.; Morriss, M.C.; Sanchez, P.; Heyne, R.; Chalak, L. Predictive Value of Neonatal MRI Showing No or Minor Degrees of Brain Injury After Hypothermia. Pediatr. Neurol. 2014, 50, 447–451. [Google Scholar] [CrossRef]
- Rutherford, M.; Ramenghi, L.A.; Edwards, A.D.; Brocklehurst, P.; Halliday, H.; Levene, M.; Strohm, B.; Thoresen, M.; Whitelaw, A.; Azzopardi, D. Assessment of brain tissue injury after moderate hypothermia in neonates with hypoxic-ischaemic encephalopathy: A nested substudy of a randomised controlled trial. Lancet Neurol. 2010, 9, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Azzopardi, D.; Strohm, B.; Marlow, N.; Brocklehurst, P.; Deierl, A.; Eddama, O.; Goodwin, J.; Halliday, H.L.; Juszczak, E.; Kapellou, O.; et al. Effects of hypothermia for perinatal asphyxia on childhood outcomes. N. Engl. J. Med. 2014, 371, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Thoresen, M.; Penrice, J.; Lorek, A.; Cady, E.B.; Wylezinska, M.; Kirkbride, V.; Cooper, C.E.; Brown, G.C.; Edwards, A.D.; Wyatt, J.S.; et al. Mild hypothermia after severe transient hypoxia-ischemia ameliorates delayed cerebral energy failure in the newborn piglet. Pediatr. Res. 1995, 37, 667–670. [Google Scholar] [CrossRef] [PubMed]
- Drury, P.P.; Gunn, E.R.; Bennet, L.; Gunn, A.J. Mechanisms of hypothermic neuroprotection. Clin. Perinatol. 2014, 41, 161–175. [Google Scholar] [CrossRef]
- Ponnusamy, V.; Yip, P.K. The role of microRNAs in newborn brain development and hypoxic ischaemic encephalopathy. Neuropharmacology 2019, 149, 55–65. [Google Scholar] [CrossRef]
- Tissier, R.; Chenoune, M.; Pons, S.; Zini, R.; Darbera, L.; Lidouren, F.; Ghaleh, B.; Berdeaux, A.; Morin, D. Mild hypothermia reduces per-ischemic reactive oxygen species production and preserves mitochondrial respiratory complexes. Resuscitation 2013, 84, 249–255. [Google Scholar] [CrossRef]
- Leaw, B.; Nair, S.; Lim, R.; Thornton, C.; Mallard, C.; Hagberg, H. Mitochondria, Bioenergetics and Excitotoxicity: New Therapeutic Targets in Perinatal Brain Injury. Front. Cell. Neurosci. 2017, 11, 199. [Google Scholar] [CrossRef]
- Silachev, D.; Plotnikov, E.; Zorova, L.; Pevzner, I.; Sumbatyan, N.; Korshunova, G.; Gulyaev, M.; Pirogov, Y.; Skulachev, V.; Zorov, D. Neuroprotective Effects of Mitochondria-Targeted Plastoquinone and Thymoquinone in a Rat Model of Brain Ischemia/Reperfusion Injury. Molecules 2015, 20, 14487–14503. [Google Scholar] [CrossRef]
- Andrzejewski, S.; Gravel, S.-P.; Pollak, M.; St-Pierre, J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. 2014, 2, 12. [Google Scholar] [CrossRef]
- Carey, B.W.; Finley, L.W.S.; Cross, J.R.; Allis, C.D.; Thompson, C.B. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2014, 518, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Desai, N.; Roman, A.; Rochelson, B.; Gupta, M.; Xue, X.; Chatterjee, P.K.; Tam Tam, H.; Metz, C.N. Maternal metformin treatment decreases fetal inflammation in a rat model of obesity and metabolic syndrome. Am. J. Obstet. Gynecol. 2013, 209, e131–e136. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zou, L.; Jack, C.R.; Yang, Y.; Yang, E.S. Neuroprotective effect of Coenzyme Q10 on ischemic hemisphere in aged mice with mutations in the amyloid precursor protein. Neurobiol. Aging 2007, 28, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Belousova, M.; Tokareva, O.G.; Gorodetskaya, E.; Kalenikova, E.I.; Medvedev, O.S. Intravenous Treatment with Coenzyme Q10 Improves Neurological Outcome and Reduces Infarct Volume After Transient Focal Brain Ischemia in Rats. J. Cardiovasc. Pharmacol. 2016, 67, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Kalayci, M.; Unal, M.M.; Gul, S.; Acikgoz, S.; Kandemir, N.; Hanci, V.; Edebali, N.; Acikgoz, B. Effect of Coenzyme Q10 on ischemia and neuronal damage in an experimental traumatic brain-injury model in rats. BMC Neurosci. 2011, 12, 75. [Google Scholar] [CrossRef]
- Reddy, P.H. Mitochondrial Oxidative Damage in Aging and Alzheimer′s Disease: Implications for Mitochondrially Targeted Antioxidant Therapeutics. BioMed Res. Int. 2006, 2006, 31372. [Google Scholar] [CrossRef]
- Papazisis, G.; Pourzitaki, C.; Sardeli, C.; Lallas, A.; Amaniti, E.; Kouvelas, D. Deferoxamine decreases the excitatory amino acid levels and improves the histological outcome in the hippocampus of neonatal rats after hypoxia-ischemia. Pharmacol. Res. 2008, 57, 73–78. [Google Scholar] [CrossRef]
- Battelli, M.G.; Polito, L.; Bortolotti, M.; Bolognesi, A. Xanthine Oxidoreductase-Derived Reactive Species: Physiological and Pathological Effects. Oxid. Med. Cell Longev. 2016, 2016, 3527579. [Google Scholar] [CrossRef]
- Kelley, E.E. A new paradigm for XOR-catalyzed reactive species generation in the endothelium. Pharmacol. Rep. 2015, 67, 669–674. [Google Scholar] [CrossRef]
- Casetta, B.; Longini, M.; Proietti, F.; Perrone, S.; Buonocore, G. Development of a fast and simple LC-MS/MS method for measuring the F2-isoprostanes in newborns. J. Matern. Fetal Neonatal Med. 2012, 25, 114–118. [Google Scholar] [CrossRef]
- Van Bel, F.; Shadid, M.; Moison, R.M.; Dorrepaal, C.A.; Fontijn, J.; Monteiro, L.; Van De Bor, M.; Berger, H.M. Effect of allopurinol on postasphyxial free radical formation, cerebral hemodynamics, and electrical brain activity. Pediatrics 1998, 101, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Benders, M.J.; Bos, A.F.; Rademaker, C.M.; Rijken, M.; Torrance, H.L.; Groenendaal, F.; van Bel, F. Early postnatal allopurinol does not improve short term outcome after severe birth asphyxia. Arch. Dis. Child. Fetal Neonatal Ed. 2006, 91, F163–F165. [Google Scholar] [CrossRef] [PubMed]
- Garg, B.; Sharma, D.; Bansal, A. Systematic review seeking erythropoietin role for neuroprotection in neonates with hypoxic ischemic encephalopathy: Presently where do we stand. J. Matern. Fetal Neonatal Med. 2018, 31, 3214–3224. [Google Scholar] [CrossRef] [PubMed]
- Robertson, N.J.; Tan, S.; Groenendaal, F.; van Bel, F.; Juul, S.E.; Bennet, L.; Derrick, M.; Back, S.A.; Chavez Valdez, R.; Northington, F.; et al. Which Neuroprotective Agents are Ready for Bench to Bedside Translation in the Newborn Infant? J. Pediatr. 2012, 160, 544–552.e544. [Google Scholar] [CrossRef] [PubMed]
- McAdams, R.M.; Juul, S.E. Neonatal Encephalopathy: Update on Therapeutic Hypothermia and Other Novel Therapeutics. Clin. Perinatol. 2016, 43, 485–500. [Google Scholar] [CrossRef]
- Alonso-Alconada, D.; Álvarez, A.; Arteaga, O.; Martínez-Ibargüen, A.; Hilario, E. Neuroprotective Effect of Melatonin: A Novel Therapy against Perinatal Hypoxia-Ischemia. Int. J. Mol. Sci. 2013, 14, 9379–9395. [Google Scholar] [CrossRef]
- Carloni, S.; Perrone, S.; Buonocore, G.; Longini, M.; Proietti, F.; Balduini, W. Melatonin protects from the long-term consequences of a neonatal hypoxic-ischemic brain injury in rats. J. Pineal Res. 2007, 44, 157–164. [Google Scholar] [CrossRef]
- Elbini Dhouib, I.; Jallouli, M.; Annabi, A.; Gharbi, N.; Elfazaa, S.; Lasram, M.M. A minireview on N-acetylcysteine: An old drug with new approaches. Life Sci. 2016, 151, 359–363. [Google Scholar] [CrossRef]
- Jenkins, D.D.; Wiest, D.B.; Mulvihill, D.M.; Hlavacek, A.M.; Majstoravich, S.J.; Brown, T.R.; Taylor, J.J.; Buckley, J.R.; Turner, R.P.; Rollins, L.G.; et al. Fetal and Neonatal Effects of N-Acetylcysteine When Used for Neuroprotection in Maternal Chorioamnionitis. J. Pediatr. 2016, 168, 67–76.e66. [Google Scholar] [CrossRef]
- Sekhon, B.; Sekhon, C.; Khan, M.; Patel, S.J.; Singh, I.; Singh, A.K. N-Acetyl cysteine protects against injury in a rat model of focal cerebral ischemia. Brain Res. 2003, 971, 1–8. [Google Scholar] [CrossRef]
- Nie, X.; Lowe, D.W.; Rollins, L.G.; Bentzley, J.; Fraser, J.L.; Martin, R.; Singh, I.; Jenkins, D. Sex-specific effects of N-acetylcysteine in neonatal rats treated with hypothermia after severe hypoxia-ischemia. Neurosci. Res. 2016, 108, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Shin, K.; Choi, E.-K.; Choi, Y.; Jang, J.-Y.; Kim, J.; Jeong, H.-S.; Lee, W.; Lee, Y.-B.; Kim, S.U.; et al. Protective Effects ofN-Acetyl-L-Cysteine in Human Oligodendrocyte Progenitor Cells and Restoration of Motor Function in Neonatal Rats with Hypoxic-Ischemic Encephalopathy. Evid.-Based Complement. Altern. Med. 2015, 2015, 764251. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, X.; Mao, Z.Y.; Wang, X.M.; Liu, Z.L. Neuroprotective effect of docosahexaenoic acid on glutamate-induced cytotoxicity in rat hippocampal cultures. Neuroreport 2003, 14, 2457–2461. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Yanai, H.; Namiki, Y.; Fukatsu-Sasaki, K.; Furutani, N.; Tada, N. Neuroprotective effects of edaravone: A novel free radical scavenger in cerebrovascular injury. CNS Drug Rev. 2006, 12, 9–20. [Google Scholar] [CrossRef]
- Gathwala, G.; Marwah, A.; Gahlaut, V.; Marwah, P. Effect of high-dose phenobarbital on oxidative stress in perinatal asphyxia: An open label randomized controlled trial. Indian. Pediatr. 2011, 48, 613–617. [Google Scholar] [CrossRef]
- Domoki, F.; Olah, O.; Zimmermann, A.; Nemeth, I.; Toth-Szuki, V.; Hugyecz, M.; Temesvari, P.; Bari, F. Hydrogen is neuroprotective and preserves cerebrovascular reactivity in asphyxiated newborn pigs. Pediatr. Res. 2010, 68, 387–392. [Google Scholar] [CrossRef]
- Peeters-Scholte, C.; Braun, K.; Koster, J.; Kops, N.; Blomgren, K.; Buonocore, G.; van Buul-Offers, S.; Hagberg, H.; Nicolay, K.; van Bel, F.; et al. Effects of Allopurinol and Deferoxamine on Reperfusion Injury of the Brain in Newborn Piglets after Neonatal Hypoxia-Ischemia. Pediatr. Res. 2003, 54, 516–522. [Google Scholar] [CrossRef]
- Laptook, A.R. Birth Asphyxia and Hypoxic-Ischemic Brain Injury in the Preterm Infant. Clin. Perinatol. 2016, 43, 529–545. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, Y.; Li, T.; Wang, X.; Zhu, C. Iron Metabolism and Brain Development in Premature Infants. Front. Physiol. 2019, 10, 463. [Google Scholar] [CrossRef]
- Rathnasamy, G.; Ling, E.A.; Kaur, C. Iron and iron regulatory proteins in amoeboid microglial cells are linked to oligodendrocyte death in hypoxic neonatal rat periventricular white matter through production of proinflammatory cytokines and reactive oxygen/nitrogen species. J. Neurosci. 2011, 31, 17982–17995. [Google Scholar] [CrossRef]
- Emerit, J.; Beaumont, C.; Trivin, F. Iron metabolism, free radicals, and oxidative injury. Biomed. Pharmacother. 2001, 55, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.K.; Labhasetwar, V. Nanoparticle-mediated delivery of superoxide dismutase to the brain: An effective strategy to reduce ischemia-reperfusion injury. FASEB J. 2009, 23, 1384–1395. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, J.; Zhao, F.; Wang, H.; Qu, Y.; Mu, D. Nitric oxide synthase in hypoxic or ischemic brain injury. Rev. Neurosci. 2015, 26, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Favie, L.M.A.; Cox, A.R.; van den Hoogen, A.; Nijboer, C.H.A.; Peeters-Scholte, C.; van Bel, F.; Egberts, T.C.G.; Rademaker, C.M.A.; Groenendaal, F. Nitric Oxide Synthase Inhibition as a Neuroprotective Strategy Following Hypoxic-Ischemic Encephalopathy: Evidence From Animal Studies. Front. Neurol. 2018, 9, 258. [Google Scholar] [CrossRef]
- Li, J.; Yawno, T.; Sutherland, A.; Loose, J.; Nitsos, I.; Bischof, R.; Castillo-Melendez, M.; McDonald, C.A.; Wong, F.Y.; Jenkin, G.; et al. Preterm white matter brain injury is prevented by early administration of umbilical cord blood cells. Exp. Neurol. 2016, 283, 179–187. [Google Scholar] [CrossRef]
- Paton, M.C.B.; McDonald, C.A.; Allison, B.J.; Fahey, M.C.; Jenkin, G.; Miller, S.L. Perinatal Brain Injury As a Consequence of Preterm Birth and Intrauterine Inflammation: Designing Targeted Stem Cell Therapies. Front. Neurosci. 2017, 11, 200. [Google Scholar] [CrossRef]
- Zhou, W.; Fu, Y.; Zhang, M.; Buabeid, M.A.; Ijaz, M.; Murtaza, G. Nanoparticle-mediated therapy of neuronal damage in the neonatal brain. J. Drug Deliv. Sci. Technol. 2021, 61, 102208. [Google Scholar] [CrossRef]
- Khan, I.; Saeed, K.; Khan, I. Nanoparticles: Properties, applications and toxicities. Arab. J. Chem. 2019, 12, 908–931. [Google Scholar] [CrossRef]
- Khan, A.R.; Yang, X.; Fu, M.; Zhai, G. Recent progress of drug nanoformulations targeting to brain. J. Control. Release 2018, 291, 37–64. [Google Scholar] [CrossRef]
- Kim, J.; Mirando, A.C.; Popel, A.S.; Green, J.J. Gene delivery nanoparticles to modulate angiogenesis. Adv. Drug Deliv. Rev. 2017, 119, 20–43. [Google Scholar] [CrossRef]
- Liao, R.; Wood, T.R.; Nance, E. Nanotherapeutic modulation of excitotoxicity and oxidative stress in acute brain injury. Nanobiomedicine 2020, 7, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, J.; Shamenkov, D.; Petrov, V.; Ramge, P.; Cychutek, K.; Koch-Brandt, C.; Alyautdin, R. Apolipoprotein-mediated Transport of Nanoparticle-bound Drugs Across the Blood-Brain Barrier. J. Drug Target. 2008, 10, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Yellepeddi, V.K.; Joseph, A.; Nance, E. Pharmacokinetics of nanotechnology-based formulations in pediatric populations. Adv. Drug Deliv. Rev. 2019, 151–152, 44–55. [Google Scholar] [CrossRef]
- Nowak, M.; Brown, T.D.; Graham, A.; Helgeson, M.E.; Mitragotri, S. Size, shape, and flexibility influence nanoparticle transport across brain endothelium under flow. Bioeng. Transl. Med. 2019, 5, e10153. [Google Scholar] [CrossRef]
- Zhang, C.; Nance, E.A.; Mastorakos, P.; Chisholm, J.; Berry, S.; Eberhart, C.; Tyler, B.; Brem, H.; Suk, J.S.; Hanes, J. Convection enhanced delivery of cisplatin-loaded brain penetrating nanoparticles cures malignant glioma in rats. J. Control. Release 2017, 263, 112–119. [Google Scholar] [CrossRef]
- Curtis, C.; Zhang, M.; Liao, R.; Wood, T.; Nance, E. Systems-level thinking for nanoparticle-mediated therapeutic delivery to neurological diseases. WIREs Nanomed. Nanobiotechnol. 2016, 9, e1422. [Google Scholar] [CrossRef]
- Liao, R.; Wood, T.R.; Nance, E. Superoxide dismutase reduces monosodium glutamate-induced injury in an organotypic whole hemisphere brain slice model of excitotoxicity. J. Biol. Eng. 2020, 14, 3. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Item | Description |
|---|---|
| Search date | 31 August 2024 |
| Time frame | From initiation to 31 August 2024 |
| Search terms | ‘Oxidate stress’, ‘oxidants’, ‘antioxidants’, ‘free radicals’ and ‘perinatal asphyxia’, ‘hypoxic-ischemic encephalopathy’, ‘neonatal encephalopathy’ |
| Database | Pubmed |
| Inclusion and exclusion criteria | English, full-text articles |
| Biomarker | Mechanism of Action | Fluid to Measure | Phase of Hypoxemia/Ischemia |
|---|---|---|---|
| Xanthine oxidase | Converts hypoxanthine and xanthine to an oxygen radical-producing form of xanthine dehydrogenase | Blood | Hypoxia/ischemia |
| Superoxide dismutase | Increases acutely to defend against ROS | Blood, CSF | Hypoxia/ischemia |
| Glutathione peroxidase | Increases acutely to defend against ROS | Blood, CSF | Hypoxia/ischemia |
| Catalase | Increases acutely to defend against ROS | Blood, CSF | Hypoxia/ischemia |
| Nitric oxide | Increases due to the upregulation of NOS during ischemia | Blood, CSF | Hypoxia/ischemia |
| Perfusion-assisted breathing | Assay to evaluate oxidative stress | Blood | Hypoxia/ischemia |
| Malondialdehyde | Biomarker of damage to n-3 and n-6 fatty acids | Blood, CSF | Hypoxia/ischemia followed by reperfusion/reoxygenation |
| 4-hydroxynonenal | Byproduct of damage to n-6 fatty acids | Blood | Hypoxia/ischemia followed by reperfusion/reoxygenation |
| Prostaglandin-like compounds (isoprostanes, neuroprostanes, neurofurans) | Formed from the free radical-induced breakdown of arachidonic acid and docosahexaenoic acid | Blood | Hypoxia/ischemia followed by reperfusion/reoxygenation |
| Protein oxidation markers (protein carbonyls) | Formed through protein carbonylation, nitration, crosslinking, or the loss of thiol groups by free radicals | Blood | Hypoxia/ischemia followed by reperfusion/reoxygenation |
| Uric acid | Increased lately during the development of oxidative damage | Urine | Reperfusion/reoxygenation |
| Iron not-bound by proteins | Iron interacts with O2− and H2O2 forming highly reactive OH | Blood | Reperfusion/reoxygenation |
| Option | Mechanism of Action | Phase of Hypoxemia/Ischemia |
|---|---|---|
| Hypothermia | Decreases ROS Decreases brain metabolism Controls activity of glial cells Prevents cell death | Within six hours of hypoxia/ischemia Reperfusion phase |
| Mitochondrial therapy: | ||
| 1. Metformin. | Suppresses mitochondrial complex I in the respiratory chain Affects inflammatory T cells | Hypoxia/ischemia phase Reperfusion phase |
| 2. Coenzyme Q10. | Decreases the production of ROS | Hypoxia/ischemia phase Reperfusion phase |
| 3. Mitoquinone. | Reduces superoxide production, lipid peroxidation, and ROS generation | Hypoxia/ischemia phase Reperfusion phase |
| Antioxidants | ||
| 1. Free radical initiators and scavengers: | ||
| I. Allopurinol. | Inhibits xanthine oxidase Chelator for non-protein-bound iron | Reperfusion phase |
| II. Erythropoietin. | Reduces inflammation Decreases ROS Decreases caspase activation Prevents cell death | Hypoxia/ischemia phase Reperfusion phase |
| III. Melatonin. | Antioxidant, anti-inflammatory, and anti-apoptotic action | Hypoxia/ischemia phase Reperfusion phase |
| IV. N-acetylcysteine. | Antioxidant (neutralizes ROS) Reduces the severity of hypoxic–ischemic damage Reduces the extent of demyelination in the corpus callosum Enhances myelin production | Hypoxia/ischemia phase Reperfusion phase |
| V. Docosahexaenoic acid. | Antioxidant (neutralizes ROS) Reduces glutamate excitotoxicity Decreases nitric oxide levels Enhances antioxidant enzyme activities | Hypoxia/ischemia phase Reperfusion phase |
| VI. Edaravone. | Antioxidant (neutralizes ROS) | Reperfusion phase |
| VII. Phenobarbital. | Reduces the levels of lipid peroxides and antioxidant enzymes | Within two hours of hypoxia/ischemia Reperfusion phase |
| VIII. Molecular hydrogen. | Antioxidant Maintains the reactivity of blood vessels | Hypoxia/ischemia phase Reperfusion phase |
| IX. Deferoxamine. | Removes iron excess Decrease excitatory amino acids Increases VEGF and erythropoietin | Hypoxia/ischemia phase Reperfusion phase |
| X. Superoxide dismutase. | Antioxidant (neutralizes ROS) | Reperfusion phase |
| 2. Nitric oxide inhibitors. | Maintains cerebral blood flow | Hypoxia/ischemia phase |
| 3. Magnesium. | Blocks NMDA receptors Reduces excessive calcium or glutamate Reduces inflammation Reduces oxidative stress | Hypoxia/ischemia phase Reperfusion phase |
| Stem cells | Reduce inflammation | Hypoxia/ischemia phase Reperfusion phase |
| Nanomaterials | Targeted therapeutic material | Hypoxia/ischemia phase Reperfusion phase |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rallis, D.; Dermitzaki, N.; Baltogianni, M.; Kapetaniou, K.; Giapros, V. Balance of Antioxidants vs. Oxidants in Perinatal Asphyxia. Appl. Sci. 2024, 14, 9651. https://doi.org/10.3390/app14219651
Rallis D, Dermitzaki N, Baltogianni M, Kapetaniou K, Giapros V. Balance of Antioxidants vs. Oxidants in Perinatal Asphyxia. Applied Sciences. 2024; 14(21):9651. https://doi.org/10.3390/app14219651
Chicago/Turabian StyleRallis, Dimitrios, Niki Dermitzaki, Maria Baltogianni, Konstantina Kapetaniou, and Vasileios Giapros. 2024. "Balance of Antioxidants vs. Oxidants in Perinatal Asphyxia" Applied Sciences 14, no. 21: 9651. https://doi.org/10.3390/app14219651
APA StyleRallis, D., Dermitzaki, N., Baltogianni, M., Kapetaniou, K., & Giapros, V. (2024). Balance of Antioxidants vs. Oxidants in Perinatal Asphyxia. Applied Sciences, 14(21), 9651. https://doi.org/10.3390/app14219651