
Pharmacoinformatics, Molecular Dynamics Simulation, and Quantum Mechanics Calculation Based Phytochemical Screening of Croton bonplandianum Against Breast Cancer by Targeting Estrogen Receptor-α (ERα)

 ,
,  , , ,
, , ,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Compounds Retrieval and Preparation
2.2. Pharmacokinetics Properties of Compounds
2.3. Thermodynamic Properties of the Compounds
2.4. FMO of the Compounds
2.5. Molecular Docking and Post-Docking Visualization
2.6. Molecular Dynamics Simulation (MDS)
2.6.1. Simulation Trajectory Analysis
2.6.2. RMSD
2.6.3. RMSF
3. Results
3.1. Construction of a Ligands Library
3.2. Pharmacokinetics Analysis
3.3. Analysis of Thermodynamic Properties of the Phyto-Compounds
3.4. FMO Analysis of the Compounds
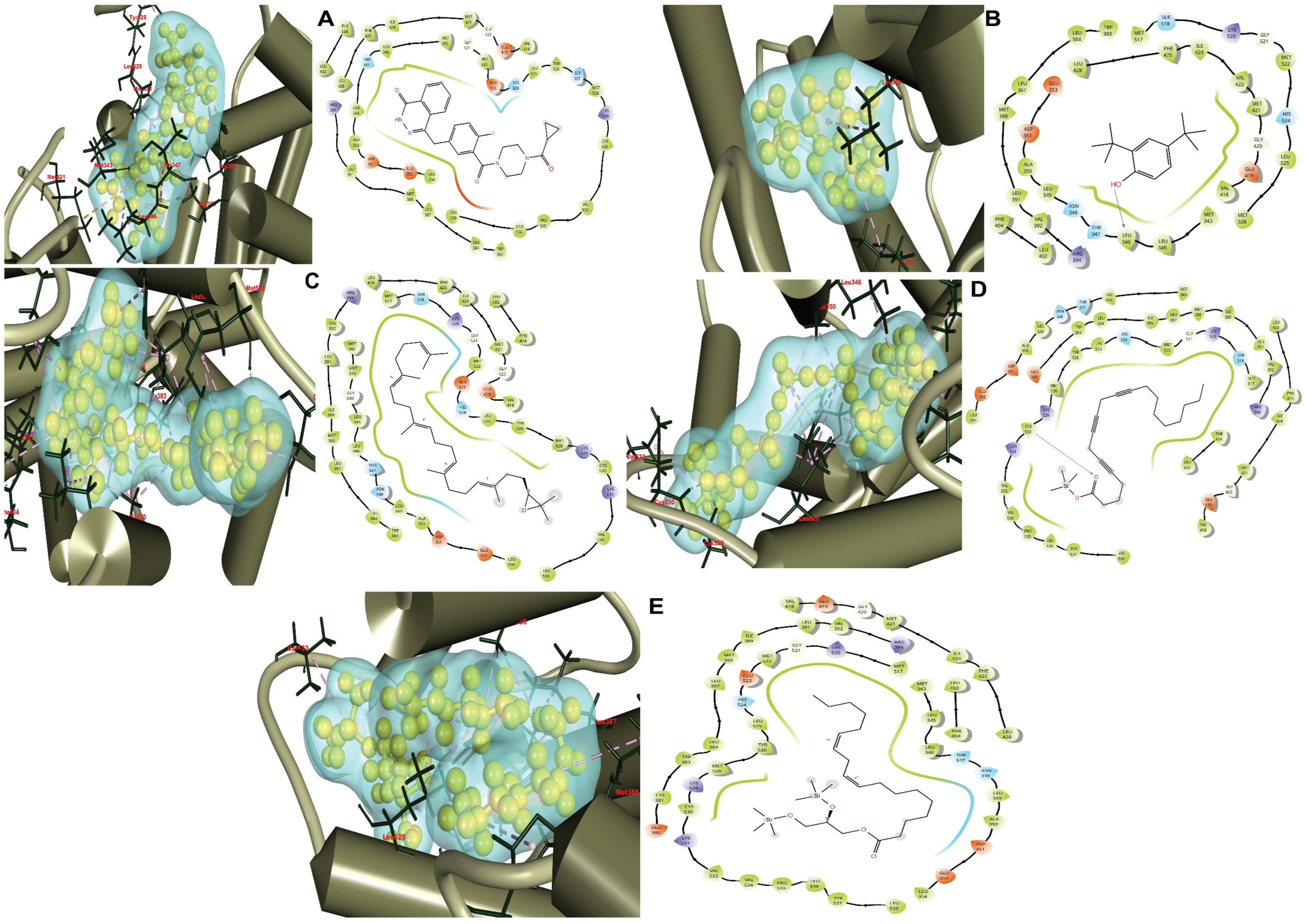
3.5. Site-Specific Molecular Docking
3.6. Protein–Ligands Interactions Analysis
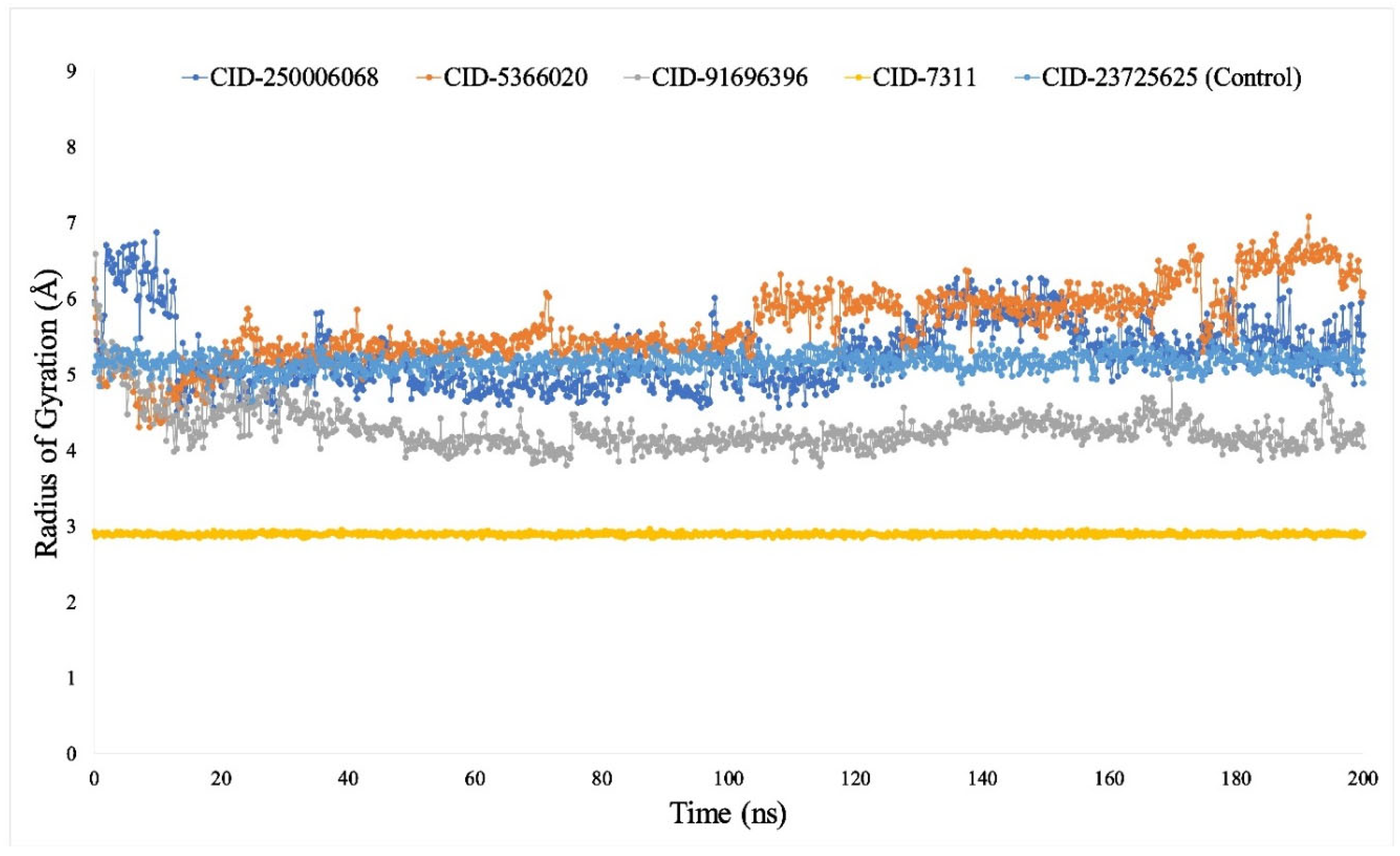
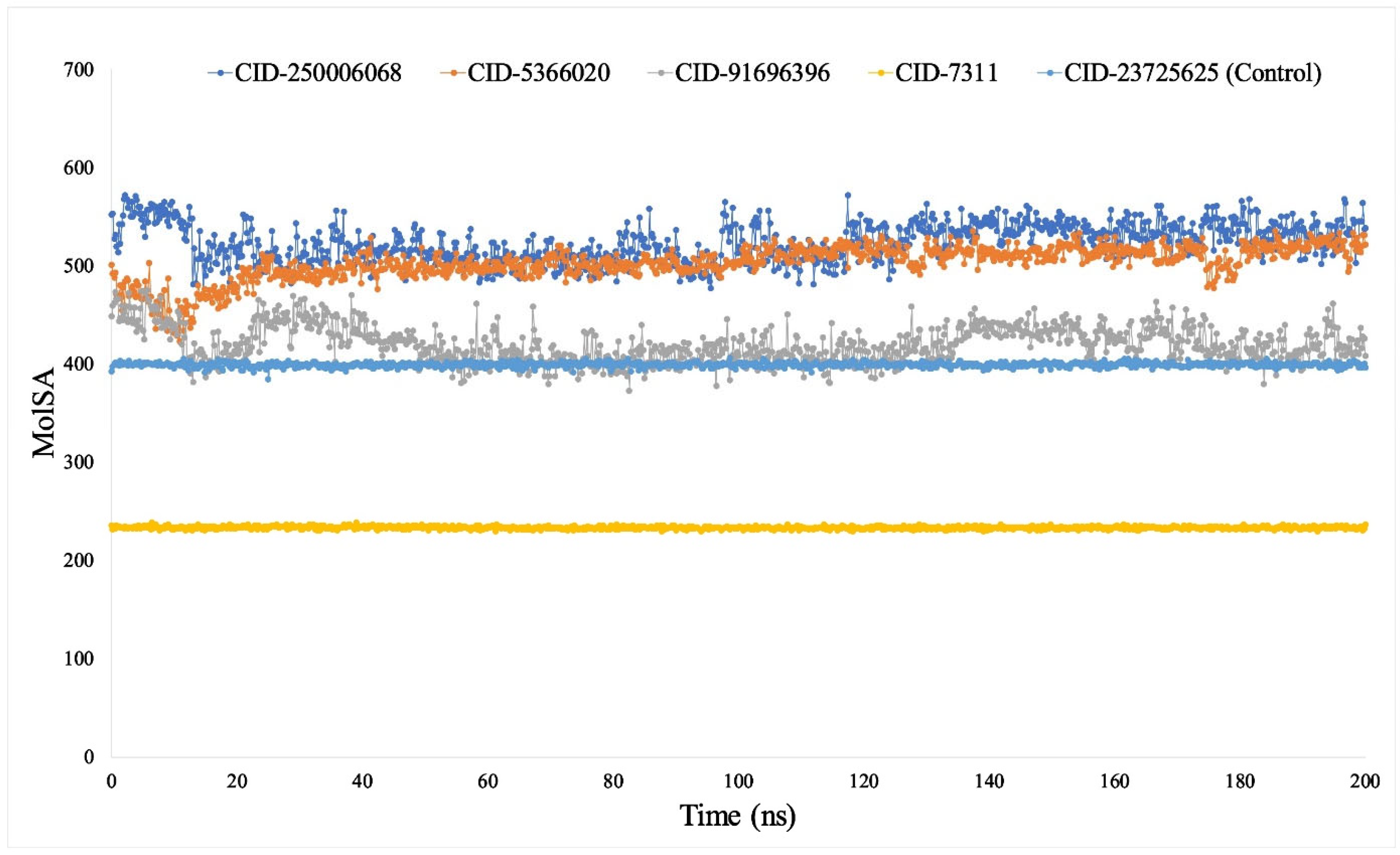
3.7. Analysis of MDS Trajectories
3.7.1. RMSD Analysis
3.7.2. RMSF Analysis
3.7.3. Rg Analysis
3.7.4. Analysis of SASA, MolSA and PSA
3.7.5. Analysis of Intramolecular Bonds
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sharma, R. Breast cancer incidence, mortality and mortality-to-incidence ratio (MIR) are associated with human development, 1990–2016: Evidence from Global Burden of Disease Study 2016. Breast Cancer 2019, 26, 428–445. [Google Scholar] [CrossRef] [PubMed]
- Sohel, M.; Biswas, P.; Al Amin, M.; Hossain, M.A.; Sultana, H.; Dey, D.; Aktar, S.; Setu, A.; Khan, M.S.; Paul, P.; et al. Genistein, a Potential Phytochemical Against Breast Cancer Treatment-Insight into the Molecular Mechanisms. Processes 2022, 10, 415. [Google Scholar] [CrossRef]
- Abd El-Rehim, D.M.; Pinder, S.E.; Paish, C.E.; Bell, J.; Blamey, R.W.; Robertson, J.F.; Nicholson, R.I.; Ellis, I.O. Expression of luminal and basal cytokeratins in human breast carcinoma. J. Pathol. 2004, 203, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Youlden, D.R.; Cramb, S.M.; Dunn, N.A.; Muller, J.M.; Pyke, C.M.; Baade, P.D. The descriptive epidemiology of female breast cancer: An international comparison of screening, incidence, survival and mortality. Cancer Epidemiol. 2012, 36, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Al Saber, M.; Biswas, P.; Dey, D.; Kaium, M.A.; Islam, M.A.; Tripty, M.I.A.; Rahman, M.H.; Rahaman, T.I.; Biswas, M.Y.; Paul, P.; et al. A Comprehensive Review of Recent Advancements in Cancer Immunotherapy and Generation of CAR T Cell by CRISPR-Cas9. Processes 2022, 10, 16. [Google Scholar] [CrossRef]
- Baral, S.K.; Biswas, P.; Kaium, M.A.; Islam, M.A.; Dey, D.; Saber, M.A.; Rahaman, T.I.; Emran, T.B.; Hasan, M.N.; Jeong, M.K.; et al. A Comprehensive Discussion in Vaginal Cancer Based on Mechanisms, Treatments, Risk Factors and Prevention. Front. Oncol. 2022, 12, 883805. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Breast Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/breast-cancer#:~:text=In%202020%2C%20there%20were%202.3,the%20world’s%20most%20prevalent%20cancer (accessed on 12 July 2023).
- Shah, A.N.; Gradishar, W.J. Adjuvant Anthracyclines in Breast Cancer: What Is Their Role? Oncologist 2018, 23, 1153–1161. [Google Scholar] [CrossRef]
- Rivera, E.; Gomez, H. Chemotherapy resistance in metastatic breast cancer: The evolving role of ixabepilone. Breast Cancer Res. BCR 2010, 12 (Suppl. 2), S2. [Google Scholar] [CrossRef]
- Wan, X.; Hou, J.; Liu, S.; Zhang, Y.; Li, W.; Zhang, Y.; Ding, Y. Estrogen Receptor α Mediates Doxorubicin Sensitivity in Breast Cancer Cells by Regulating E-Cadherin. Front. Cell Dev. Biol. 2021, 9, 583572. [Google Scholar] [CrossRef]
- Payne, S.J.; Bowen, R.L.; Jones, J.L.; Wells, C.A. Predictive markers in breast cancer—The present. Histopathology 2008, 52, 82–90. [Google Scholar] [CrossRef]
- Fox, E.M.; Davis, R.J.; Shupnik, M.A. ERbeta in breast cancer—Onlooker, passive player, or active protector? Steroids 2008, 73, 1039–1051. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.R. Integration of the extranuclear and nuclear actions of estrogen. Mol. Endocrinol. 2005, 19, 1951–1959. [Google Scholar] [CrossRef] [PubMed]
- Narod, S.A. Hormone replacement therapy and the risk of breast cancer. Nat. Rev. Clin. Oncol. 2011, 8, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Al Azad, S.; Ahmed, S.; Biswas, P.; Mia, M.A.R.; Farjana, M.; Arshe, F.A.; Mily, S.J.; Ankhi, A.B.; Shaikat, M.M.; Sultana, S.; et al. Quantitative analysis of the factors influencing IDA and TSH downregulation in correlation to the fluctuation of activated vitamin D3 in women. JABET 2022, 5, 320–333. [Google Scholar] [CrossRef]
- Khan, A.M.; Sharif, M.A.; Salekeen, R.; Rahman, M.H.; Mahmud, S.; Biswas, P.; Hasan, M.N.; Islam, K.M.D.; Billah, M.M.; Islam, M.E. In vitro and in silico investigation of garlic’s (Allium sativum) bioactivity against 15-lipoxygenase mediated inflammopathies. J. Herbmed Pharmacol. 2023, 12, 283–298. [Google Scholar] [CrossRef]
- Sanyakamdhorn, S.; Agudelo, D.; Bekale, L.; Tajmir-Riahi, H.A. Targeted conjugation of breast anticancer drug tamoxifen and its metabolites with synthetic polymers. Colloids Surf. B Biointerfaces 2016, 145, 55–63. [Google Scholar] [CrossRef]
- Chang, M. Tamoxifen resistance in breast cancer. Biomol. Ther. 2012, 20, 256–267. [Google Scholar] [CrossRef]
- Subarnas, A.; Diantini, A.; Abdulah, R.; Zuhrotun, A.; Hadisaputri, Y.E.; Puspitasari, I.M.; Yamazaki, C.; Kuwano, H.; Koyama, H. Apoptosis induced in MCF-7 human breast cancer cells by 2′,4′-dihydroxy-6-methoxy-3,5-dimethylchalcone isolated from Eugenia aquea Burm f. leaves. Oncol. Lett. 2015, 9, 2303–2306. [Google Scholar] [CrossRef]
- Cohen, L.H.; Remley, M.J.; Raunig, D.; Vaz, A.D. In vitro drug interactions of cytochrome p450: An evaluation of fluorogenic to conventional substrates. Drug Metab. Dispos. 2003, 31, 1005–1015. [Google Scholar] [CrossRef]
- Society, A.C. Cancer Facts & Figures; American Cancer Society: Atlanta, GA, USA, 2016. [Google Scholar]
- Elkum, N.; Dermime, S.; Ajarim, D.; Al-Zahrani, A.; Alsayed, A.; Tulbah, A.; Al Malik, O.; Alshabanah, M.; Ezzat, A.; Al-Tweigeri, T. Being 40 or younger is an independent risk factor for relapse in operable breast cancer patients: The Saudi Arabia experience. BMC Cancer 2007, 7, 222. [Google Scholar] [CrossRef]
- Younas, M.; Hano, C.; Giglioli-Guivarc’h, N.; Abbasi, B.H. Mechanistic evaluation of phytochemicals in breast cancer remedy: Current understanding and future perspectives. RSC Adv. 2018, 8, 29714–29744. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Schmitz, J.C.; Lin, X.; Tai, N.; Yan, W.; Farrell, M.; Bailly, M.; Chen, T.; Chu, E. Thymidylate synthase as a translational regulator of cellular gene expression. Biochim. Biophys. Acta 2002, 1587, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Chaudhuri, T.K. Pharmacological aspect of Croton bonplandianus Baill: A comprehensive review. J. Pharmacogn. Phytochem. 2018, 7, 811–813. [Google Scholar]
- Vijayamuthuramalingam, U.D.K.; Rajaram, R.; Kuppusamy, K.M.; Jonnalagadda, B.; Arokiasamy, S. Anti-hyperglycemic and antioxidant potential of Croton bonplandianus Bail fractions in correlation with polyphenol content. Iran. J. Basic Med. Sci. 2017, 20, 1390–1397. [Google Scholar] [CrossRef]
- Jeeshna, M.; Paulsamy, S.; Mallikadevi, T.J. Phytochemical constituents and antimicrobial studies of the exotic plant species, Croton bonplandianum Baill. J. Life Sci. 2011, 3, 23–27. [Google Scholar] [CrossRef]
- Bhavana, J.; Kalaivani, M.K.; Sumathy, A. Cytotoxic and pro-apoptotic activities of leaf extract of Croton bonplandianus Baill. against lung cancer cell line A549. Indian J. Exp. Biol. 2016, 54, 379–385. [Google Scholar]
- Lanchhana, D.S.; Ranjit, M.; Kumar, M.S. Anticancer Activity Study of Some Selected Indian Medicinal Plants Used Traditionally. J. Pharm. Negat. Results 2023, 14, 1123–1132. [Google Scholar] [CrossRef]
- Islam, M.; Rahman, M.; Rahman, M.; Qayum, M.; Alam, M. In vitro evaluation of Croton bonplandianum Baill. as potential antitumor properties using Agrobacterium tumefaciens. J. Agric. Technol. 2010, 6, 79–86. [Google Scholar]
- Mahmud, S.; Uddin, M.A.R.; Paul, G.K.; Shimu, M.S.S.; Islam, S.; Rahman, E.; Islam, A.; Islam, M.S.; Promi, M.M.; Emran, T.B.; et al. Virtual screening and molecular dynamics simulation study of plant-derived compounds to identify potential inhibitors of main protease from SARS-CoV-2. Brief. Bioinform. 2021, 22, 1402–1414. [Google Scholar] [CrossRef]
- Biswas, P.; Dey, D.; Rahman, A.; Islam, M.A.; Susmi, T.F.; Kaium, M.A.; Hasan, M.N.; Rahman, M.D.H.; Mahmud, S.; Saleh, M.A.; et al. Analysis of SYK Gene as a Prognostic Biomarker and Suggested Potential Bioactive Phytochemicals as an Alternative Therapeutic Option for Colorectal Cancer: An In-Silico Pharmaco-Informatics Investigation. J. Pers. Med. 2021, 11, 888. [Google Scholar] [CrossRef]
- Biswas, P.; Hasan, M.M.; Dey, D.; Dos Santos Costa, A.C.; Polash, S.A.; Bibi, S.; Ferdous, N.; Kaium, M.A.; Rahman, M.D.H.; Jeet, F.K.; et al. Candidate antiviral drugs for COVID-19 and their environmental implications: A comprehensive analysis. Environ. Sci. Pollut. Res. Int. 2021, 28, 59570–59593. [Google Scholar] [CrossRef] [PubMed]
- Andalib, K.M.S.; Biswas, P.; Sakib, M.R.; Hasan, M.N.; Rahman, M.H.; Habib, A. Identification of novel MCM2 inhibitors from Catharanthus roseus by pharmacoinformatics, molecular docking and molecular dynamics simulation-based evaluation. Inform. Med. Unlocked 2023, 39, 101251. [Google Scholar] [CrossRef]
- Ahmed, S.S.; Rahman, M.O.; Alqahtani, A.S.; Sultana, N.; Almarfadi, O.M.; Ali, M.A.; Lee, J. Anticancer potential of phytochemicals from Oroxylum indicum targeting Lactate Dehydrogenase A through bioinformatic approach. Toxicol. Rep. 2023, 10, 56–75. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Liu, C.; Wang, Q.; Lin, P.; Cheng, F. In silico polypharmacology of natural products. Brief. Bioinform. 2017, 19, 1153–1171. [Google Scholar] [CrossRef]
- Ahammad, F.; Tengku Abd Rashid, T.R.; Mohamed, M.; Tanbin, S.; Ahmad Fuad, F.A. Contemporary Strategies and Current Trends in Designing Antiviral Drugs Against Dengue Fever via Targeting Host-Based Approaches. Microorganisms 2019, 7, 296. [Google Scholar] [CrossRef]
- Lu, S.; Ji, M.; Ni, D.; Zhang, J. Discovery of hidden allosteric sites as novel targets for allosteric drug design. Drug Discov. Today 2018, 23, 359–365. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef]
- Singh, H.; Bharadvaja, N. Treasuring the computational approach in medicinal plant research. Prog. Biophys. Mol. Biol. 2021, 164, 19–32. [Google Scholar] [CrossRef]
- Mohanraj, K.; Karthikeyan, B.S.; Vivek-Ananth, R.P.; Chand, R.P.B.; Aparna, S.R.; Mangalapandi, P.; Samal, A. IMPPAT: A curated database of Indian Medicinal Plants, Phytochemistry And Therapeutics. Sci. Rep. 2018, 8, 4329. [Google Scholar] [CrossRef]
- Hou, T.; Xu, X. ADME evaluation in drug discovery. 3. Modeling blood-brain barrier partitioning using simple molecular descriptors. J. Chem. Inf. Comput. Sci. 2003, 43, 2137–2152. [Google Scholar] [CrossRef]
- Aribisala, J.O.; S’thebe, N.W.; Sabiu, S. In silico exploration of phenolics as modulators of penicillin binding protein (PBP) 2× of Streptococcus pneumoniae. Sci. Rep. 2024, 14, 8788. [Google Scholar] [CrossRef]
- Bhatia, M.J.C.T. An overview of conceptual-DFT based insights into global chemical reactivity of volatile sulfur compounds (VSCs). Comput. Toxicol. 2023, 29, 100295. [Google Scholar] [CrossRef]
- Klein, J.; Pilmé, J. Exploring the Reactivity of Donor–Acceptor Systems through a Combined Conceptual and Constrained DFT Approach. J. Chem. Theory Comput. 2024, 20, 2010–2021. [Google Scholar] [CrossRef]
- Koltai, P.; Kunde, P. A Koopman–Takens theorem: Linear least squares prediction of nonlinear time series. Commun. Math. Phys. 2024, 405, 120. [Google Scholar] [CrossRef]
- Uzzaman, M.; Hoque, M.J. Physiochemical, molecular docking, and pharmacokinetic studies of Naproxen and its modified derivatives based on DFT. Int. J. Sci. Res. Manag. 2018, 6, 2018–2025. [Google Scholar] [CrossRef]
- Siddiquee, N.H.; Talukder, M.E.K.; Ahmed, E.; Zeba, L.T.; Aivy, F.S.; Rahman, M.H.; Barua, D.; Rumman, R.; Hossain, M.I.; Shimul, M.E.K.; et al. Cheminformatics-based analysis identified (Z)-2-(2,5-dimethoxy benzylidene)-6-(2-(4-methoxyphenyl)-2-oxoethoxy) benzofuran-3(2H)-one as an inhibitor of Marburg replication by interacting with NP. Microb. Pathog. 2024, 195, 106892. [Google Scholar] [CrossRef]
- Yuan, X.-Q.; Yu, X.-H.; Zhu, X.-L.; Wang, X.-C.; Liu, X.-Y.; Cao, J.-W.; Qin, X.-L.; Zhang, P. Comparative Analysis of the Hydrogen Bond Vibrations of Ice XII. ACS Omega 2022, 7, 2970–2974. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; pp. 84–es. [Google Scholar]
- Morshed, A.; Al Azad, S.; Mia, M.A.R.; Uddin, M.F.; Ema, T.I.; Yeasin, R.B.; Srishti, S.A.; Sarker, P.; Aurthi, R.Y.; Jamil, F.; et al. Oncoinformatic screening of the gene clusters involved in the HER2-positive breast cancer formation along with the in silico pharmacodynamic profiling of selective long-chain omega-3 fatty acids as the metastatic antagonists. Mol. Divers. 2022, 27, 2651–2672. [Google Scholar] [CrossRef]
- Paul, P.K.; Al Azad, S.; Rahman, M.H.; Farjana, M.; Uddin, M.R.; Dey, D.; Mahmud, S.; Ema, T.I.; Biswas, P.; Anjum, M.; et al. Catabolic profiling of selective enzymes in the saccharification of non-food lignocellulose parts of biomass into functional edible sugars and bioenergy: An in silico bioprospecting. J. Adv. Vet. Anim. Res. 2022, 9, 19–32. [Google Scholar] [CrossRef]
- Rahman, M.D.H.; Biswas, P.; Dey, D.; Hannan, M.A.; Sahabuddin, M.; Araf, Y.; Kwon, Y.; Emran, T.B.; Ali, M.S.; Uddin, M.J. An In-Silico Identification of Potential Flavonoids Against Kidney Fibrosis Targeting TGFβR-1. Life 2022, 12, 1764. [Google Scholar] [CrossRef]
- Jian, J.; He, D.; Gao, S.; Tao, X.; Dong, X. Pharmacokinetics in Pharmacometabolomics: Towards Personalized Medication. Pharmaceuticals 2023, 16, 1568. [Google Scholar] [CrossRef] [PubMed]
- Vora, L.K.; Gholap, A.D.; Jetha, K.; Thakur, R.R.S.; Solanki, H.K.; Chavda, V.P. Artificial Intelligence in Pharmaceutical Technology and Drug Delivery Design. Pharmaceutics 2023, 15, 1916. [Google Scholar] [CrossRef] [PubMed]
- Shaker, B.; Ahmad, S.; Lee, J.; Jung, C.; Na, D. In silico methods and tools for drug discovery. Comput. Biol. Med. 2021, 137, 104851. [Google Scholar] [CrossRef] [PubMed]
- Wadood, A.; Ahmed, N.; Shah, L.; Ahmad, A.; Hassan, H.; Shams, S. In-silico drug design: An approach which revolutionarised the drug discovery process. OA Drug Des. Deliv. 2013, 1, 3. [Google Scholar]
- Gupta, M.; Kant, K.; Sharma, R.; Kumar, A. Evaluation of In Silico Anti-parkinson Potential of β-asarone. Cent. Nerv. Syst. Agents Med. Chem. 2018, 18, 128–135. [Google Scholar] [CrossRef]
- Pasha, M.A.; Mondal, S.; Panigrahi, N.; Shetye, G.; Ma, R.; Franzblau, S.G.; Zheng, Y.T.; Murugesan, S. One-Pot Synthesis of Novel Hydrazono-1,3-Thıazolıdın-4-One Derivatives as Anti-HIV and Anti-Tubercular Agents: Synthesıs, Bıologıcal Evaluatıon, Molecular Modelling and Admet Studıes. Curr. HIV Res. 2022, 20, 255–271. [Google Scholar] [CrossRef]
- Abdullah, A.; Biswas, P.; Sahabuddin, M.; Mubasharah, A.; Khan, D.A.; Hossain, A.; Roy, T.; Rafi, N.M.R.; Dey, D.; Hasan, M. Molecular Dynamics Simulation and Pharmacoinformatic Integrated Analysis of Bioactive Phytochemicals from Azadirachta indica (Neem) to Treat Diabetes Mellitus. J. Chem. 2023, 2023, 4170703. [Google Scholar] [CrossRef]
- Aziz, S.; Bibi, S.; Hasan, M.M.; Biswas, P.; Ali, M.I.; Bilal, M.; Chopra, H.; Mukerjee, N.; Maitra, S. A review on influence of biochar amendment on soil processes and environmental remediation. Biotechnol. Genet. Eng. Rev. 2023, 6, 1–35. [Google Scholar] [CrossRef]
- Ferdausi, N.; Islam, S.; Rimti, F.H.; Quayum, S.T.; Arshad, E.M.; Ibnat, A.; Islam, T.; Arefin, A.; Ema, T.I.; Biswas, P.; et al. Point-specific interactions of isovitexin with the neighboring amino acid residues of the hACE2 receptor as a targeted therapeutic agent in suppressing the SARS-CoV-2 influx mechanism. J. Adv. Vet. Anim. Res. 2022, 9, 230–240. [Google Scholar] [CrossRef]
- Krupanidhi, S.; Abraham Peele, K.; Venkateswarulu, T.; Ayyagari, V.S.; Nazneen Bobby, M.; John Babu, D.; Venkata Narayana, A.; Aishwarya, G. Screening of phytochemical compounds of Tinospora cordifolia for their inhibitory activity on SARS-CoV-2: An in silico study. J. Biomol. Struct. Dyn. 2021, 39, 5799–5803. [Google Scholar] [CrossRef]
- Elfiky, A.A.; Elshemey, W.M. Molecular dynamics simulation revealed binding of nucleotide inhibitors to ZIKV polymerase over 444 nanoseconds. J. Med. Virol. 2018, 90, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, S.; Rahman, E.; Nain, Z.; Billah, M.; Karmakar, S.; Mohanto, S.C.; Paul, G.K.; Amin, A.; Acharjee, U.K.; Saleh, M.A. Computational discovery of plant-based inhibitors against human carbonic anhydrase IX and molecular dynamics simulation. J. Biomol. Struct. Dyn. 2021, 39, 2754–2770. [Google Scholar] [CrossRef] [PubMed]
- Alamri, M.A.; Altharawi, A.; Alabbas, A.B.; Alossaimi, M.A.; Alqahtani, S.M. Structure-based virtual screening and molecular dynamics of phytochemicals derived from Saudi medicinal plants to identify potential COVID-19 therapeutics. Arab. J. Chem. 2020, 13, 7224–7234. [Google Scholar] [CrossRef] [PubMed]
- Kousar, K.; Majeed, A.; Yasmin, F.; Hussain, W.; Rasool, N. Phytochemicals from selective plants have promising potential against SARS-CoV-2: Investigation and corroboration through molecular docking, MD simulations, and quantum computations. BioMed Res. Int. 2020, 2020, 6237160. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Mwe (g/mol) | HBA | HBD | Num Rot | T.P.S.A. (Å2) | Log P | S.B. | LD50 | HpT | AT | MToD | Lp. Violation | ToC |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CID-23725625 (Control) | 434.46 | 5 | 1 | 6 | 86.37 | 2.78 | 0.55 | 2.623 | Yes | No | 0.204 | 0 | 0.569 |
| CID-7311 | 206.32 | 1 | 1 | 2 | 20.23 | 3.99 | 0.55 | 2.351 | No | No | 0.421 | 0 | 0.759 |
| CID-5366020 | 426.72 | 1 | 0 | 15 | 12.53 | 8.55 | 0.55 | 1.622 | No | No | −0.319 | 1 | 1.396 |
| CID-91696396 | 372.62 | 2 | 0 | 11 | 26.30 | 6.40 | 0.55 | 1.874 | No | No | −0.571 | 1 | 1.702 |
| CID-250006068 | 498.89 | 4 | 0 | 22 | 44.76 | 8.41 | - | 2.262 | No | No | −0.097 | 1 | 1.598 |
| Name | Stoichiometry | Electronic Energy (Hartree) | Enthalpy (Hartree) | Gibbs Free Energy (Hartree) | Dipole Moment (Debye) |
|---|---|---|---|---|---|
| CID-23725625 (Control) | C24H23FN4O3 | −685.636 | −685.635 | −685.687 | 2.889 |
| CID-7311 | C14H22O | −621.660 | −621.659 | −621.719 | 1.335 |
| CID-5366020 | C30H50O | −1247.741 | −1247.740 | −1247.859 | 1.859 |
| CID-91696396 | C23H36O2Si | −1337.485 | −1337.484 | −1337.591 | 1.601 |
| CID-250006068 | C27H54O4Si2 | −1025.885 | −1025.565 | −1025.873 | 2.014 |
| Molecules (Chair) | εHOMO | εLUMO | Gap | H (Hardness, Gap/2) | S (Softness, 1/Hardness) |
|---|---|---|---|---|---|
| 23725625 (Control) | −0.207 | −0.002 | 0.205 | 0.102 | 9.803 |
| 7311 | −0.208 | −0.005 | 0.203 | 0.101 | 9.900 |
| 5366020 | −0.215 | −0.002 | 0.213 | 0.106 | 9.433 |
| 91696396 | −0.239 | −0.008 | 0.231 | 0.115 | 8.695 |
| 250006068 | −0.314 | −0.005 | 0.309 | 0.154 | 6.493 |
| Complex | Bonding Energy (kcal/mol) | H-Bond | Hydrophobic | Polar | Positive | Negative |
|---|---|---|---|---|---|---|
| olaparib (CID-23725625) (control drug) | −7.9 | PHE 404, LEU402, LEU428, PHE425, ILE424, MET421, MET343, LEU346, LEU349, ALA350, LEU354, TRP383, LEU384, LEU387, MET388, LEU391, VAL418, GLY420, GLY521, MET522, LEU525, TYR526, MET528, CYS530, VAL533, PRO535, LEU536, LEU539 | THR347 HIS524 SER527 | ARG394 LYS529 | ASP351 GLU353 GLU419 GLU523 | |
| 2,4-di-tert-butylphenol (CID-7311) | −7.8 | LEU346 | MET343, LEU345, LEU346, LEU349, ALA350, MET388, LEU387, LEU384, TRP383, LEU391, VAL392, LEU402, PHE404, VAL418, GLY420, MET421, VAL422, ILE424, PHE425, LEU428, MET517, GLY521, MET522, LEU525, MET528 | ASN348 THR347 SER518 HIS524 | ARG399 LYS520 | GLU353 ASP351 GLU419 |
| 2,3-oxidosqualene (CID-5366020) | −8.6 | MET342, MET343, GLY344, LEU345, LEU346, LEU349, ALA350, LEU354, TRP383, LEU384, LEU387, MET388, ILE389, LEU391, VAL392, LEU402, PHE404, VAL418, GLY420, MET421, ILE424, PHE425, LEU428, MET517, GLY521, MET522, LEU525, TYR526, MET528, CYS530, VAL533, LEU536 | THR347 ASN348 SER518 HIS524 | ARG394 LYS520 LYS529 LYS531 | ASP351 GLU353 GLU419 GLU523 | |
| 5,8,11-eicosatriynoic acid, Trimethylsilyl Ester (CID-91696396) | −8.2 | Cys530 | MET343, LEU346, LEU349, ALA350, LEU354, TRP383, LEU384, ILE386, LEU387, MET388, ILE389, LEU391, VAL392, VAL418, GLY420, MET421, ILE424, PHE425, LEU428, MET517, GLY521, MET522, LEU525, TYR526, MET528, CYS530, VAL533, VAL534, PRO535, LEU536, TYR537, LEU539 | THR347 ASN348 SER518 HIS524 | ARG394 LYS520 LYS529 LYS531 | ASP351 GLU353 GLU380 GLU419 |
| 1-monolinolein (CID-250006068) | −8.2 | MET343, LEU345, LEU346, LEU349, ALA350, LEU354, CYS381, TRP383, LEU384, LEU387, MET388, ILE389, LEU391, VAL392, VAL418, GLY420, MET421, ILE424, PHE425, LEU428, MET517, GLY521, MET522, LEU525, TYR526, MET528, CYS530, VAL533, VAL534, PRO535, LEU536, TYR537, LEU539 | THR347 ASN348 HIS524 | ARG394 LYS520 LYS529 LYS531 | ASP351 GLU353 GLU380 GLU419 GLU523 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saha, S.; Biswas, P.; Tareq, M.I.; Rahman Sakib, M.; Akter Rakhi, S.; Zilani, M.N.H.; Harrath, A.H.; Rahman, M.A.; Hasan, M.N. Pharmacoinformatics, Molecular Dynamics Simulation, and Quantum Mechanics Calculation Based Phytochemical Screening of Croton bonplandianum Against Breast Cancer by Targeting Estrogen Receptor-α (ERα). Appl. Sci. 2024, 14, 9878. https://doi.org/10.3390/app14219878
Saha S, Biswas P, Tareq MI, Rahman Sakib M, Akter Rakhi S, Zilani MNH, Harrath AH, Rahman MA, Hasan MN. Pharmacoinformatics, Molecular Dynamics Simulation, and Quantum Mechanics Calculation Based Phytochemical Screening of Croton bonplandianum Against Breast Cancer by Targeting Estrogen Receptor-α (ERα). Applied Sciences. 2024; 14(21):9878. https://doi.org/10.3390/app14219878
Chicago/Turabian StyleSaha, Shuvo, Partha Biswas, Mohaimenul Islam Tareq, Musfiqur Rahman Sakib, Suraia Akter Rakhi, Md. Nazmul Hasan Zilani, Abdel Halim Harrath, Md. Ataur Rahman, and Md. Nazmul Hasan. 2024. "Pharmacoinformatics, Molecular Dynamics Simulation, and Quantum Mechanics Calculation Based Phytochemical Screening of Croton bonplandianum Against Breast Cancer by Targeting Estrogen Receptor-α (ERα)" Applied Sciences 14, no. 21: 9878. https://doi.org/10.3390/app14219878
APA StyleSaha, S., Biswas, P., Tareq, M. I., Rahman Sakib, M., Akter Rakhi, S., Zilani, M. N. H., Harrath, A. H., Rahman, M. A., & Hasan, M. N. (2024). Pharmacoinformatics, Molecular Dynamics Simulation, and Quantum Mechanics Calculation Based Phytochemical Screening of Croton bonplandianum Against Breast Cancer by Targeting Estrogen Receptor-α (ERα). Applied Sciences, 14(21), 9878. https://doi.org/10.3390/app14219878