

Understanding Polymers Through Transfer Learning and Explainable AI


Abstract
:1. Introduction
2. Methods
2.1. Datasets
2.2. Data Treatment
2.3. ANN Architecture
2.4. Training and Optimization
2.5. Transfer Learning and Comparison
2.6. Shapley Value Analysis for Model Interpretability
3. Results and Discussion
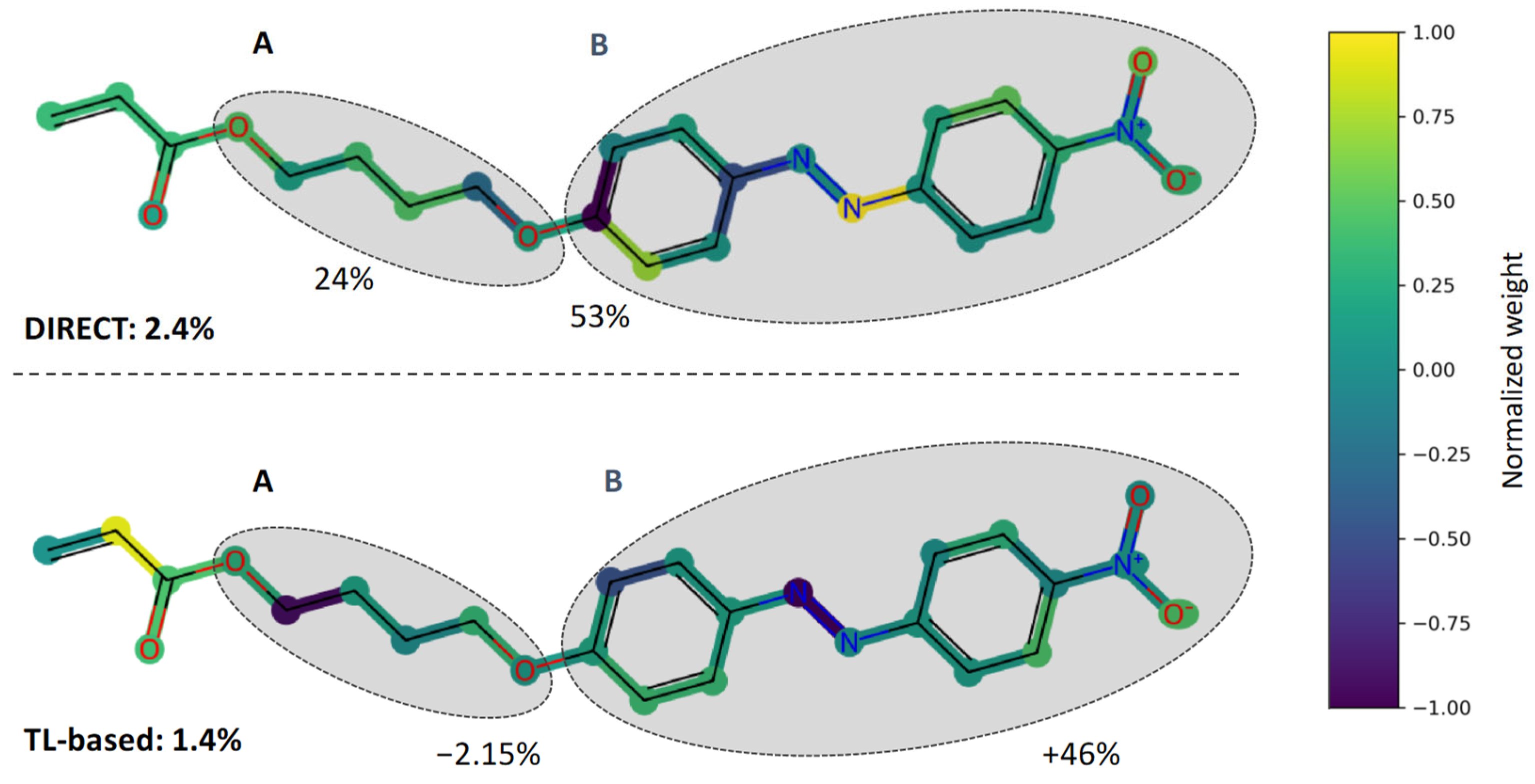
3.1. Direct Modelling of the System
3.2. Modelling of the System from Transfer Learning on Pre Trained Model
3.3. Comparison of Both Approaches
4. Conclusions
Supplementary Materials
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rai, A. Explainable AI: From black box to glass box. J. Acad. Mark. Sci. 2020, 48, 137–141. [Google Scholar] [CrossRef]
- Hassija, V.; Chamola, V.; Mahapatra, A.; Singal, A.; Goel, D.; Huang, K.; Scardapane, S.; Spinelli, I.; Mahmud, M.; Hussain, A. Interpreting Black-Box Models: A Review on Explainable Artificial Intelligence. Cogn. Comput. 2024, 16, 45–74. [Google Scholar] [CrossRef]
- Selvaraju, R.R.; Cogswell, M.; Das, A.; Vedantam, R.; Parikh, D.; Batra, D. Grad-CAM: Visual Explanations from Deep Networks via Gradient-Based Localization. Int. J. Comput. Vis. 2020, 128, 336–359. [Google Scholar] [CrossRef]
- Hu, J.; Li, Z.; Lin, J.; Zhang, L. Prediction and Interpretability of Glass Transition Temperature of Homopolymers by Data-Augmented Graph Convolutional Neural Networks. ACS Appl. Mater. Interfaces 2023, 15, 54006–54017. [Google Scholar] [CrossRef]
- Miccio, L.A.; Schwartz, G.A. Localizing and quantifying the intra-monomer contributions to the glass transition temperature using artificial neural networks. Polymer 2020, 203, 122786. [Google Scholar] [CrossRef]
- Borredon, C.; Miccio, L.A.; Cerveny, S.; Schwartz, G.A. Characterising the glass transition temperature-structure relationship through a recurrent neural network. J. Non-Cryst. Solids X 2023, 18, 100185. [Google Scholar] [CrossRef]
- Liu, J.; Wu, Y.; Lin, Z.; Peng, L.; Chu, Q.; Tang, Y.; Zhang, W. Visual analytics of an interpretable prediction model for the glass transition temperature of fluoroelastomers. Mater. Today Commun. 2024, 40, 110155. [Google Scholar] [CrossRef]
- Montavon, G.; Samek, W.; Müller, K.R. Methods for interpreting and understanding deep neural networks. Digit. Signal Process. 2018, 73, 1–15. [Google Scholar] [CrossRef]
- Oviedo, F.; Ferres, J.L.; Buonassisi, T.; Butler, K.T. Interpretable and explainable machine learning for materials science and chemistry. Acc. Mater. Res. 2022, 3, 597–607. [Google Scholar] [CrossRef]
- Roscher, R.; Bohn, B.; Duarte, M.F.; Garcke, J. Explainable machine learning for scientific insights and discoveries. IEEE Access 2020, 8, 42200–42216. [Google Scholar] [CrossRef]
- Zhong, X.; Gallagher, B.; Liu, S.; Kailkhura, B.; Hiszpanski, A.; Han, T.Y.-J. Explainable machine learning in materials science. npj Comput. Mater. 2022, 8, 204. [Google Scholar] [CrossRef]
- Adadi, A.; Berrada, M. Peeking inside the black-box: A survey on explainable artificial intelligence (xai). IEEE Access 2018, 6, 52138–52160. [Google Scholar] [CrossRef]
- Fiosina, J.; Sievers, P.; Drache, M.; Beuermann, S. Polymer reaction engineering meets explainable machine learning. Comput. Chem. Eng. 2023, 177, 108356. [Google Scholar] [CrossRef]
- Arrieta, A.B.; Díaz-Rodríguez, N.; Del Ser, J.; Bennetot, A.; Tabik, S.; Barbado, A.; Garcia, S.; Gil-Lopez, S.; Molina, D.; Benjamins, R.; et al. Explainable artificial intelligence (xai): Concepts, taxonomies, opportunities and challenges toward responsible ai. Inf. Fusion 2020, 58, 82–115. [Google Scholar] [CrossRef]
- Vilone, G.; Longo, L. Notions of explainability and evaluation approaches for explainable artificial intelligence. Inf. Fusion 2021, 76, 89–106. [Google Scholar] [CrossRef]
- Chen, G.; Tao, L.; Li, Y. Predicting Polymers’ Glass Transition Temperature by a Chemical Language Processing Model. Polymers 2021, 13, 1898. [Google Scholar] [CrossRef]
- Nguyen, T.; Bavarian, M. A Machine Learning Framework for Predicting the Glass Transition Temperature of Homopolymers. Ind. Eng. Chem. Res. 2022, 61, 12690–12698. [Google Scholar] [CrossRef]
- Cassar, D.R.; de Carvalho, A.C.; Zanotto, E.D. Predicting glass transition temperatures using neural networks. Acta Mater. 2018, 159, 249–256. [Google Scholar] [CrossRef]
- Mattioni, B.E.; Jurs, P.C. Prediction of Glass Transition Temperatures from Monomer and Repeat Unit Structure Using Computational Neural Networks. J. Chem. Inf. Comput. Sci. 2002, 42, 232–240. [Google Scholar] [CrossRef]
- Miccio, L.A.; Schwartz, G.A. Mapping Chemical Structure-Glass Transition Temperature Relationship through Artificial Intelligence. Macromolecules 2021, 54, 1811–1817. [Google Scholar] [CrossRef]
- Mysona, J.A.; Nealey, P.F.; de Pablo, J.J. Machine Learning Models and Dimensionality Reduction for Prediction of Polymer Properties. Macromolecules 2024, 57, 1988–1997. [Google Scholar] [CrossRef]
- Hou, F.; Wu, Z.; Hu, Z.; Xiao, Z.; Wang, L.; Zhang, X.; Li, G. Comparison Study on the Prediction of Multiple Molecular Properties by Various Neural Networks. J. Phys. Chem. A 2018, 122, 9128–9134. [Google Scholar] [CrossRef] [PubMed]
- Miccio, L.A.; Borredon, C.; Schwartz, G.A. A glimpse inside materials: Polymer structure—Glass transition temperature relationship as observed by a trained artificial intelligence. Comput. Mater. Sci. 2024, 236, 112863. [Google Scholar] [CrossRef]
- Uddin, M.J.; Fan, J. Interpretable Machine Learning Framework to Predict the Glass Transition Temperature of Polymers. Polymers 2024, 16, 1049. [Google Scholar] [CrossRef]
- Audus, D.J.; De Pablo, J.J. Polymer Informatics: Opportunities and Challenges. ACS Macro Lett. 2017, 6, 1078–1082. [Google Scholar] [CrossRef]
- Borredon, C.; Miccio, L.A.; Schwartz, G.A. Transfer learning-driven artificial intelligence model for glass transition temperature estimation of molecular glass formers mixtures. Comput. Mater. Sci. 2024, 238, 112931. [Google Scholar] [CrossRef]
- Gibbs, J.H. Nature of the Glass Transition in Polymers. J. Chem. Phys. 2004, 25, 185. [Google Scholar] [CrossRef]
- Pugar, J.A.; Childs, C.M.; Huang, C.; Haider, K.W.; Washburn, N.R. Elucidating the Physicochemical Basis of the Glass Transition Temperature in Linear Polyurethane Elastomers with Machine Learning. J. Phys. Chem. B 2020, 124, 9722–9733. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, X. Machine learning glass transition temperature of polymers. Heliyon 2020, 6, e05055. [Google Scholar] [CrossRef]
- Joyce, S.; Osguthorpe, D.; Padget, J.; Price, G. Neural network prediction of glass-transition temperatures from monomer structure. J. Chem. Soc. Faraday Trans. 1995, 91, 2491–2496. [Google Scholar] [CrossRef]
- Miccio, L.A.; Schwartz, G.A. From chemical structure to quantitative polymer properties prediction through convolutional neural networks. Polymer 2020, 193, 122341. [Google Scholar] [CrossRef]
- Volgin, I.V.; Batyr, P.A.; Matseevich, A.V.; Dobrovskiy, A.Y.; Andreeva, M.V.; Nazarychev, V.M.; Goikhman, M.Y.; Vizilter, Y.V.; Askadskii, A.A.; Lyulin, S.V. Machine Learning with Enormous ‘synthetic’ Data Sets: Predicting Glass Transition Temperature of Polyimides Using Graph Convolutional Neural Networks. ACS Omega 2022, 7, 43678–43691. [Google Scholar] [CrossRef] [PubMed]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- O’Boyle, N.M. Towards a Universal SMILES representation—A standard method to generate canonical SMILES based on the InChI. J. Cheminform. 2012, 4, 22. [Google Scholar] [CrossRef]
- Ucak, U.V.; Ashyrmamatov, I.; Lee, J. Improving the quality of chemical language model outcomes with atom-in-SMILES tokenization. J. Cheminform. 2023, 15, 55. [Google Scholar] [CrossRef]
- Wu, H.; Gu, X. Max-Pooling Dropout for Regularization of Convolutional Neural Networks. arXiv 2015, arXiv:1512.01400. [Google Scholar]
- LeCun, Y.; Kavukcuoglu, K.; Farabet, C. Convolutional networks and applications in vision. In Proceedings of the 2010 IEEE International Symposium on Circuits and Systems, Paris, France, 30 May–2 June 2010; pp. 253–256. [Google Scholar] [CrossRef]
- Kingma, D.P.; Ba, J.L. Adam: A method for stochastic optimization. In Proceedings of the 3rd International Conference on Learning Representations, ICLR 2015, San Diego, CA, USA, 7–9 May 2015. [Google Scholar]
- Sundararajan, M.; Najmi, A. The many shapley values for model explanation. In Proceedings of the 37th International Conference on Machine Learning ICML 2020, Online, 13–18 July 2020; PartF168147-12. pp. 9210–9220. [Google Scholar]
- Anjum, M.; Khan, K.; Ahmad, W.; Ahmad, A.; Amin, M.N.; Nafees, A. New SHapley Additive ExPlanations (SHAP) Approach to Evaluate the Raw Materials Interactions of Steel-Fiber-Reinforced Concrete. Materials 2022, 15, 6261. [Google Scholar] [CrossRef]
- Liu, T.; Barnard, A. Shapley Based Residual Decomposition for Instance Analysis. In Proceedings of the 40th International Conference on Machine Learning, Honolulu, HI, USA, 23–29 July 2023. [Google Scholar]
- Barnard, A.S.; Fox, B.L. Importance of Structural Features and the Influence of Individual Structures of Graphene Oxide Using Shapley Value Analysis. Chem. Mater. 2023, 35, 8840–8856. [Google Scholar] [CrossRef]
- Rubinstein, M.; Colby, R.H. Polymer Physics; Oxford University Press (OUP): Oxford, UK, 2003. [Google Scholar] [CrossRef]
- Qin, J. Similarity of Polymer Packing in Melts Is Dictated by . Macromolecules 2024, 57, 1885–1892. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aspect | Direct Model | Transfer Learning Model |
|---|---|---|
| Training data | Trained directly on polymer-specific dataset. Requires larger polymer-specific datasets. | Pre-trained on molecular dataset and fine-tuned on polymer dataset. Performs well even with small dataset due to transfer of pre-trained knowledge. |
| Prediction Accuracy | Higher accuracy for polymer-specific phenomena. | Comparable accuracy, but lower for certain polymer-specific interactions. It shows a biased output for short side chain samples. |
| Generalizability | Limited to polyacrylates (or chemically similar) data; struggles in data scarcity conditions. | Good generalization due to the use of a fingerprint that carries pre-trained knowledge; can still work under data scarcity conditions. When faced with new structures, need to adapt previous knowledge during fine tuning. As a result, in data scarcity conditions can show bias towards underrepresented samples. |
| Handling of Chain Interactions | Effectively captures polymer chain stiffness, entanglements, and intra-chain effects. | Underestimates effects of weak nonpolar interactions and intra-chain phenomena. |
| Interpretability | Shapley values indicate consistent contributions. | Shows heterogeneous Shapley contributions for short pendant chains. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miccio, L.A. Understanding Polymers Through Transfer Learning and Explainable AI. Appl. Sci. 2024, 14, 10413. https://doi.org/10.3390/app142210413
Miccio LA. Understanding Polymers Through Transfer Learning and Explainable AI. Applied Sciences. 2024; 14(22):10413. https://doi.org/10.3390/app142210413
Chicago/Turabian StyleMiccio, Luis A. 2024. "Understanding Polymers Through Transfer Learning and Explainable AI" Applied Sciences 14, no. 22: 10413. https://doi.org/10.3390/app142210413
APA StyleMiccio, L. A. (2024). Understanding Polymers Through Transfer Learning and Explainable AI. Applied Sciences, 14(22), 10413. https://doi.org/10.3390/app142210413