
Pulmonary Hypertension Secondary to Myxomatous Mitral Valve Disease in Dogs: Current Insights into the Histological Manifestation and Its Determining Factors
 , , , ,
, , , ,
Abstract
:1. Introduction
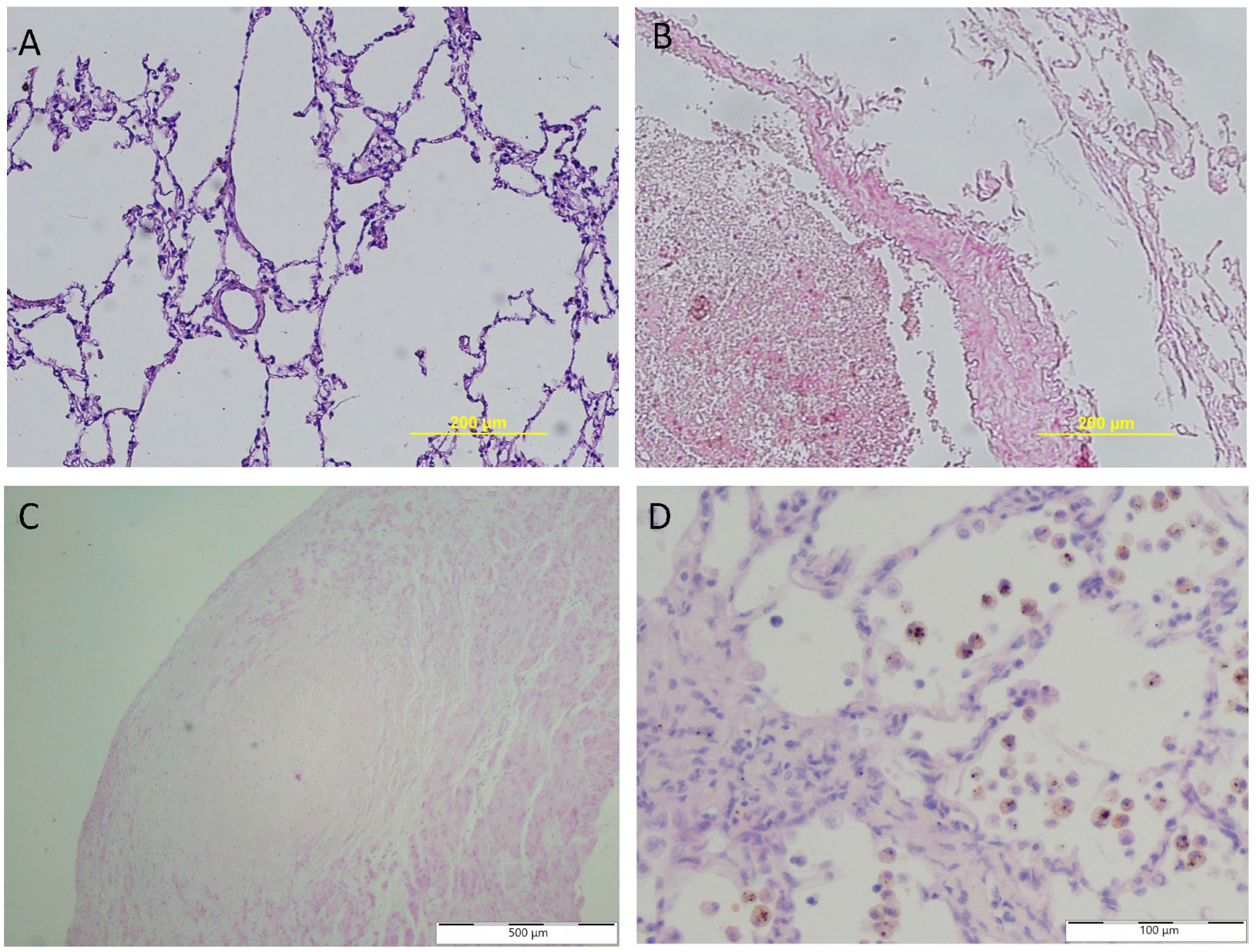
2. Histopathology in PVH Secondary to MMVD
2.1. Pulmonary Vessels

{kind=link}
| Source | Tissue | Parameter | Control Group | MMVD | MMVD + PH | ||
|---|---|---|---|---|---|---|---|
| [37] | Lung | Diameter | Internal | 266.15 ± 20.47 | 268.56 ± 20.62 | 255.92 ± 28.97 | |
| External | 299.46 ± 23.06 | 345.51 ± 28.38 | 376.76 ± 29.06 | ||||
| [43] | Lung | 20–100 μm | %MT | 13.04 ± 0.82 | 23.70 ± 1.47 | 32.44 ± 1.65 | |
| 101–200 μm | 12.60 ± 1.09 | 24.54 ± 1.45 | 30.48 ± 2.09 | ||||
| 201–300 μm | 12.77 ± 1.27 | 23.44 ± 1.28 | 30.51 ± 1.82 | ||||
| 301–400 μm | 11.71 ± 0.86 | 22.40 ± 1.58 | 32.52 ± 1.63 | ||||
| [37,44,45] | Lung | %MT | 11.04 ± 0.95 | 22.36 ± 1.44 | 32.25 ± 5.06 | ||
| [45] | Lung | SMCs | 4.87 ± 0.24 | 16.61 ± 1.91 | 23.44 ± 2.18 | ||
| [45] | Lung | SMCs | 29.04 ± 2.15 | 51.65 ± 4.11 | 69.53 ± 8.33 | ||
| [37] | Lung | %PCNA | 1.91 ± 0.21 | 14.99 ± 1.07 | 6.06 ± 0.61 | ||
2.2. Lymphatic Vessels
2.3. Pulmonary Tissue
2.4. Morphology of Blood Cells
3. Molecular Changes in PVH Secondary to MMVD
3.1. Endothelin 1
| Source | Tissue | Biomarker [Unit] | Control Group | MMVD | ||
|---|---|---|---|---|---|---|
| MMVD (PH+) | MMVD (PH−) | |||||
| Endothelin and endothelin’s receptors | ||||||
| [55] | Blood (plasma) | Et [pg/mL] | 17.8 (15.0–19.2) | 20.6 (17.2–23.1) | n/a | |
| [71] | Lung | ET [−] | n/a | 3-fold increase in the concentration | n/a | |
| [71] | Lung | ET, B [−] | n/a | 3-fold increase in the concentration | n/a | |
| Vascular endothelial growth factor and VEGF’s receptors | ||||||
| [55] | Blood (plasma) | VEGF [pg/mL] | 33.1 (29.7–36.9) | 81.2 (73.3–96.2) | n/a | |
| [71] | Lung | VEGF [−] | n/a | 3-fold increase in the concentration | n/a | |
| [71] | Lung | VEGFR [−] | n/a | 3-fold increase in the concentration | n/a | |
| Oxide nitric | ||||||
| [95] | Blood (plasma) | NO [µM] | n/a | 25.88 (15.08–36.71) | n/a | |
| Serotonin value, expression of the receptors, and associated protein | ||||||
| [55] | Blood (plasma) | SRT [ng/mL] | 26.1 (21.0–30.7) | 26.6 (22.4–30.5) | n/a | |
| [96] | Blood | plasma | SRT (ng/mL) | 2.92 (1.76–7.50) | 1.75 (1.19–2.72) | 1.23 (0.27–4.23) |
| [96] | Platelet | SRT [ng/109 platelets] | 179.73 (102.37–352.24) | 135.11 (21.21–312.22) | 325.99 (96.84–407.66) | |
| [44] | Lung | TPH1 [ppa] | 0.86 ± 0.19 | 8.07 ± 0.73 | 5.43 ± 0.34 | |
| SERT [ppa] | 0.58 ± 0.05 | 9.87 ± 0.43 | 4.56 ± 0.44 | |||
| 5HT2A [ppa] | 1.48 ± 0.11 | 7.06 ± 0.83 | 45 ± 0.46 | |||
| ERK [ppa] | 0.80 ± 0.2 | 7.78 ± 0.35 | 1.61 ± 0.47 | |||
| pERK [ppa] | 0.00 ± 0.0 | 6.61 ± 0.85 | 0.00 ± 0.00 | |||
| [97] | Lung | TPH-1 [rpe] | 2.33 (0.58–4.09) | 1.56 (0.68–5.08) | 1.59 (0.52–5.43) | |
| SERT [rpe] | 1.14 (0.09–3.10) | 1.49 (0.24–1.69) | 0.46 (0.04–1.00) | |||
| 5-HTR2A [rpe] | 4.40 (0.97–10.75) | 3.15 (2.36–8.88) | 2.59 (1.12–9.46) | |||
| ERK1/2 [rpe] | 1.74 (0.68–6.37) | 2.16 (1.04–4.29) | 1.41 (0.72–5.38) | |||
| pERK1/2 [rpe] | 0.72 (0.13–2.01) | 0.21 (0.07–2.59) | 1.02 (0.35–4.09) | |||
| Arteries | TPH-1 [rpe] | 0.65 (0.49–1.12) | 1.85 (0.51–2.95) | 0.81 (0.29–4.09) | ||
| SERT [rpe] | 0.23 (0.14–0.39) | 0.40 (0.04–0.74) | 0.14 (0.04–0.63) | |||
| 5-HTR2A [rpe] | 0.68 (0.44–1.00) | 1.58 (0.50–2.57) | 0.99 (0.37–4.67) | |||
| ERK1/2 [rpe] | 0.86 (0.73–1.04) | 1.04 (0.48–4.15) | 0.81 (0.54–4.23) | |||
| pERK1/2 [rpe] | 0.61 (0.35–0.89) | 0.56 (0.10–2.87) | 0.45 (0.11–2.26) | |||
3.2. Nitric Oxide
3.3. Serotonin
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yamini, S.H.; Jeyaraja, K.; Chandrasekhar, M.; Kavitha, S. Prevalence of Pulmonary Hypertension in Dogs with Degenerative Mitral Valve Disease. Indian J. Anim. Res. 2020, 54, 1136–1142. [Google Scholar] [CrossRef]
- DeProspero, D.J.; O’Donnell, K.A.; DeFrancesco, T.C.; Keene, B.W.; Tou, S.P.; Adin, D.B.; Atkins, C.E.; Meurs, K.M. Myxomatous Mitral Valve Disease in Miniature Schnauzers and Yorkshire Terriers: 134 Cases (2007–2016). J. Am. Vet. Med. Assoc. 2021, 259, 1428–1432. [Google Scholar] [CrossRef]
- O’Brien, M.J. Genetics and Genomics of Myxomatous Mitral Valve Disease in Dogs. Anim. Genet. 2021, 52, 409–421. [Google Scholar] [CrossRef]
- Saengklub, N.; Pirintr, P.; Nampimoon, T.; Kijtawornrat, A.; Chaiyabutr, N. Short-Term Effects of Sacubitril/Valsartan on Echocardiographic Parameters in Dogs with Symptomatic Myxomatous Mitral Valve Disease. Front. Vet. Sci. 2021, 8, 700230. [Google Scholar] [CrossRef]
- Meurs, K.M.; Adin, D.; O’Donnell, K.; Keene, B.W.; Atkins, C.E.; DeFrancesco, T.; Tou, S. Myxomatous Mitral Valve Disease in the Miniature Poodle: A Retrospective Study. Vet. J. 2019, 244, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Reimann, M.J.; Cremer, S.; Christiansen, L.; Ibragimov, E.; Gao, F.; Cirera, S.; Fredholm, M.; Olsen, L.H.; Karlskov-Mortensen, P. Mitral Valve Transcriptome Analysis in Thirty-Four Age-Matched Cavalier King Charles Spaniels with or without Congestive Heart Failure Caused by Myxomatous Mitral Valve Disease. Mamm. Genome 2023, 35, 77–89. [Google Scholar] [CrossRef]
- Serres, F.; Chetboul, V.; Tissier, R.; Sampedrano, C.C.; Gouni, V.; Nicolle, A.P.; Pouchelon, J.-L. Chordae Tendineae Rupture in Dogs with Degenerative Mitral Valve Disease: Prevalence, Survival, and Prognostic Factors (114 Cases, 2001–2006). J. Vet. Intern. Med. 2007, 21, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.-H.; Song, K.H. Assessment of Vertebral Left Atrial Size and C-Reactive Protein in Dogs with Myxomatous Mitral Valve Disease. J. Vet. Clin. 2021, 38, 16–20. [Google Scholar] [CrossRef]
- Trofimiak, R.M.; Slivinska, L.G. Diagnostic Value of Echocardiographic Indices of Left Atrial and Ventricular Morphology in Dogs with Myxomatous Mitral Valve Disease (MMVD). Ukr. J. Vet. Agric. Sci. 2021, 4, 16–23. [Google Scholar] [CrossRef]
- Ogawa, M.; Hori, Y.; Kanno, N.; Iwasa, N.; Toyohuku, T.; Isayama, N.; Yoshikawa, A.; Akabane, R.; Sakatani, A.; Miyakawa, H. Comparison of N-Terminal pro-Atrial Natriuretic Peptide and Three Cardiac Biomarkers for Discriminatory Ability of Clinical Stage in Dogs with Myxomatous Mitral Valve Disease. J. Vet. Med. Sci. 2021, 83, 705–715. [Google Scholar] [CrossRef]
- Johnson, L.R. Pulmonary Hypertension. In BSAVA Manual of Canine and Feline Cardiorespiratory Medicine; BSAVA Library: Woodrow House, UK, 2010; pp. 264–267. [Google Scholar]
- Serres, F.J.; Chetboul, V.; Tissier, R.; Sampedrano, C.C.; Gouni, V.; Nicolle, A.P.; Pouchelon, J.-L. Doppler Echocardiography–Derived Evidence of Pulmonary Arterial Hypertension in Dogs with Degenerative Mitral Valve Disease: 86 Cases (2001–2005). J. Am. Vet. Med. Assoc. 2006, 229, 1772–1778. [Google Scholar] [CrossRef]
- Sudunagunta, S.; Green, D.; Christley, R.; Dukes-McEwan, J. The Prevalence of Pulmonary Hypertension in Cavalier King Charles Spaniels Compared with Other Breeds with Myxomatous Mitral Valve Disease. J. Vet. Cardiol. 2019, 23, 21–31. [Google Scholar] [CrossRef]
- Ferasin, L.; Linney, C. Coughing in Dogs: What Is the Evidence for and against a Cardiac Cough? J. Small Anim. Pract. 2019, 60, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Ferasin, L.; Crews, L.; Biller, D.S.; Lamb, K.E.; Borgarelli, M. Risk Factors for Coughing in Dogs with Naturally Acquired Myxomatous Mitral Valve Disease. J. Vet. Intern. Med. 2013, 27, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Udomkiattikul, J.; Kirdratanasak, N.; Siritianwanitchakul, P.; Worapunyaanun, W.; Surachetpong, S.D. Factors Related to Survival Time in Dogs with Pulmonary Hypertension Secondary to Degenerative Mitral Valve Disease Stage C. Int. J. Vet. Sci. Med. 2022, 10, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Borgarelli, M.; Abbott, J.; Braz-Ruivo, L.; Chiavegato, D.; Crosara, S.; Lamb, K.; Ljungvall, I.; Poggi, M.; Santilli, R.A.; Haggstrom, J. Prevalence and Prognostic Importance of Pulmonary Hypertension in Dogs with Myxomatous Mitral Valve Disease. J. Vet. Intern. Med. 2015, 29, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Hage, R.; Petrus, L.C.; Soares, E.C.; de Toledo Barbino Sanches, M.; Hage, M.C.F.N.S. Pulmonary hypertension secondary to chronic mitral valve disease in dogs: Prevalence and echocardiographic aspects/Hipertensão arterial pulmonar secundária à doença valvar mitral crônica em cães: Prevalência e aspectos ecocardiográficos. Braz. J. Anim. Environ. Res. 2020, 3, 2210–2222. [Google Scholar] [CrossRef]
- Guglielmini, C.; Toaldo, M.B.; Chiesa, A.; Contiero, B.; Berlanda, M.; Poser, H. Effect of Temperature Variation on Hospital Admissions and Outcomes in Dogs with Myxomatous Mitral Valve Disease and New Onset Pulmonary Edema. PLoS ONE 2020, 15, e0227807. [Google Scholar] [CrossRef] [PubMed]
- Keene, B.W.; Atkins, C.E.; Bonagura, J.D.; Fox, P.R.; Häggström, J.; Fuentes, V.L.; Oyama, M.A.; Rush, J.E.; Stepien, R.; Uechi, M. ACVIM Consensus Guidelines for the Diagnosis and Treatment of Myxomatous Mitral Valve Disease in Dogs. J. Vet. Intern. Med. 2019, 33, 1127–1140. [Google Scholar] [CrossRef] [PubMed]
- Morita, T.; Harada, K.; Uechi, M. Right Heart Failure Due to Pulmonary Hypertension after Mitral Valve Repair in a Dog. J. Vet. Cardiovasc. Med. 2020, 4, 8–15. [Google Scholar] [CrossRef]
- Kellihan, H.B.; Stepien, R.L. Pulmonary Hypertension in Canine Degenerative Mitral Valve Disease. J. Vet. Cardiol. 2012, 14, 149–164. [Google Scholar] [CrossRef]
- Reinero, C.; Visser, L.C.; Kellihan, H.B.; Masseau, I.; Rozanski, E.; Clercx, C.; Williams, K.; Abbott, J.; Borgarelli, M.; Scansen, B.A. ACVIM Consensus Statement Guidelines for the Diagnosis, Classification, Treatment, and Monitoring of Pulmonary Hypertension in Dogs. J. Vet. Intern. Med. 2020, 34, 549–573. [Google Scholar] [CrossRef]
- Galiè, N.; McLaughlin, V.V.; Rubin, L.J.; Simonneau, G. An Overview of the 6th World Symposium on Pulmonary Hypertension. Eur. Respir. J. 2019, 53, 1802148. [Google Scholar] [CrossRef] [PubMed]
- Yaghi, S.; Novikov, A.; Trandafirescu, T. Clinical Update on Pulmonary Hypertension. J. Investig. Med. 2020, 68, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Rich, S. Pulmonary Hypertension. In Cardiology for the Primary Care Physician; Springer: Berlin/Heidelberg, Germany, 2001; pp. 313–318. [Google Scholar]
- Hoeper, M.M.; Ghofrani, H.-A.; Grünig, E.; Klose, H.; Olschewski, H.; Rosenkranz, S. Pulmonary Hypertension. Dtsch. Arztebl. Int. 2017, 114, 73. [Google Scholar] [CrossRef]
- Gaine, S. Pulmonary Hypertension. JAMA 2000, 284, 3160–3168. [Google Scholar] [CrossRef]
- Rubin, L.J. Primary Pulmonary Hypertension. N. Eng. J. Med. 1997, 336, 111–117. [Google Scholar] [CrossRef]
- Naeije, R.; Richter, M.J.; Rubin, L.J. The physiological basis of pulmonary arterial hypertension. Eur. Respir. J. 2022, 59. [Google Scholar] [CrossRef]
- Hoeper, M.M.; Bogaard, H.J.; Condliffe, R.; Frantz, R.; Khanna, D.; Kurzyna, M.; Langleben, D.; Manes, A.; Satoh, T.; Torres, F.; et al. Definitions and Diagnosis of Pulmonary Hypertension. J. Am. Coll. Cardiol. 2013, 62, D42–D50. [Google Scholar] [CrossRef] [PubMed]
- Vachiéry, J.-L.; Adir, Y.; Barberà, J.A.; Champion, H.; Coghlan, J.G.; Cottin, V.; De Marco, T.; Galiè, N.; Ghio, S.; Gibbs, J.S.R.; et al. Pulmonary Hypertension Due to Left Heart Diseases. J. Am. Coll. Cardiol. 2013, 62, D100–D108. [Google Scholar] [CrossRef]
- Ibe, T.; Wada, H.; Sakakura, K.; Ugata, Y.; Maki, H.; Yamamoto, K.; Seguchi, M.; Taniguchi, Y.; Jinnouchi, H.; Momomura, S.; et al. Combined Pre- and Post-Capillary Pulmonary Hypertension: The Clinical Implications for Patients with Heart Failure. PLoS ONE 2021, 16, e0247987. [Google Scholar] [CrossRef] [PubMed]
- van Duin, R.W.B.; Stam, K.; Cai, Z.; Uitterdijk, A.; Garcia-Alvarez, A.; Ibanez, B.; Danser, A.H.J.; Reiss, I.K.M.; Duncker, D.J.; Merkus, D. Transition from Post-Capillary Pulmonary Hypertension to Combined Pre- and Post-Capillary Pulmonary Hypertension in Swine: A Key Role for Endothelin. J. Physiol. 2019, 597, 1157–1173. [Google Scholar] [CrossRef] [PubMed]
- Arwood, M.J.; Vahabi, N.; Lteif, C.; Sharma, R.K.; Machado, R.F.; Duarte, J.D. Transcriptome-Wide Analysis Associates ID2 Expression with Combined Pre- and Post-Capillary Pulmonary Hypertension. Sci. Rep. 2019, 9, 19572. [Google Scholar] [CrossRef]
- Lee, J.; Mizuno, M.; Mizuno, T.; Harada, K.; Uechi, M. Pathologic Manifestations on Surgical Biopsy and Their Correlation with Clinical Indices in Dogs with Degenerative Mitral Valve Disease. J. Vet. Intern. Med. 2015, 29, 1313–1321. [Google Scholar] [CrossRef]
- Sakarin, S.; Rungsipipat, A.; Surachetpong, S.D. Histopathological Changes of Pulmonary Vascular Remodeling in Dogs Affected with Pulmonary Hypertension Secondary to Degenerative Mitral Valve Disease. J. Vet. Cardiol. 2021, 36, 141–152. [Google Scholar] [CrossRef]
- Liu, Y.; Zhu, X.-Y.; Song, Z.-L.; Qin, M.; Xu, C.-H.; Liu, X. Right atrial mechanism contributes to atrial fibrillation in a canine model of pulmonary arterial hypertension. bioRxiv 2022, 2022-02. [Google Scholar]
- Omote, K.; Sorimachi, H.; Obokata, M.; Reddy, Y.N.V.; Verbrugge, F.H.; Omar, M.; DuBrock, H.M.; Redfield, M.M.; Borlaug, B.A. Pulmonary Vascular Disease in Pulmonary Hypertension Due to Left Heart Disease: Pathophysiologic Implications. Eur. Heart J. 2022, 43, 3417–3431. [Google Scholar] [CrossRef]
- Bacakova, L.; Travnickova, M.; Filova, E.; Matějka, R.; Stepanovska, J.; Musilkova, J.; Zarubova, J.; Molitor, M. The role of vascular smooth muscle cells in the physiology and pathophysiology of blood vessels. In Muscle Cell and Tissue-Current Status of Research Field; InTech Open: Rijeka, Croatia, 2018; pp. 229–257. [Google Scholar]
- Kellihan, H.B.; Stepien, R.L. Pulmonary Hypertension in Dogs: Diagnosis and Therapy. Vet. Clin. N. Am. Small Anim. Pract. 2010, 40, 623–641. [Google Scholar] [CrossRef]
- Moraes, D.L.; Colucci, W.S.; Givertz, M.M. Secondary Pulmonary Hypertension in Chronic Heart Failure: The Role of the Endothelium in Pathophysiology and Management. Circulation 2000, 102, 1718–1723. [Google Scholar] [CrossRef]
- Sakarin, S.; Rungsipipat, A.; Surachetpong, S.D. Perivascular Inflammatory Cells and Their Association with Pulmonary Arterial Remodelling in Dogs with Pulmonary Hypertension Due to Myxomatous Mitral Valve Disease. Vet. Res. Commun. 2023, 47, 1505–1521. [Google Scholar] [CrossRef] [PubMed]
- Sakarin, S.; Surachetpong, S.D.; Rungsipipat, A. The Expression of Proteins Related to Serotonin Pathway in Pulmonary Arteries of Dogs Affected with Pulmonary Hypertension Secondary to Degenerative Mitral Valve Disease. Front. Vet. Sci. 2020, 7, 612130. [Google Scholar] [CrossRef]
- Sakarin, S.; Rungsipipat, A.; Surachetpong, S.D. Expression of Apoptotic Proteins in the Pulmonary Artery of Dogs with Pulmonary Hypertension Secondary to Degenerative Mitral Valve Disease. Res. Vet. Sci. 2022, 145, 238–247. [Google Scholar] [CrossRef]
- Delgado, J.F.; Conde, E.; Sánchez, V.; López-Ríos, F.; Gómez-Sánchez, M.A.; Escribano, P.; Sotelo, T.; de la Cámara, A.G.; Cortina, J.; de la Calzada, C.S. Pulmonary Vascular Remodeling in Pulmonary Hypertension Due to Chronic Heart Failure. Eur. J. Heart Fail. 2005, 7, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- Chazova, I.; Robbins, I.; Loyd, J.; Newman, J.; Tapson, V.; Zhdaov, V.; Meyrick, B. Venous and Arterial Changes in Pulmonary Veno-Occlusive Disease, Mitral Stenosis and Fibrosing Mediastinitis. Eur. Respir. J. 2000, 15, 116–122. [Google Scholar] [CrossRef]
- Savai, R.; Pullamsetti, S.S.; Kolbe, J.; Bieniek, E.; Voswinckel, R.; Fink, L.; Scheed, A.; Ritter, C.; Dahal, B.K.; Vater, A.; et al. Immune and Inflammatory Cell Involvement in the Pathology of Idiopathic Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Zabka, T.S.; Campbell, F.E.; Wilson, D.W. Pulmonary Arteriopathy and Idiopathic Pulmonary Arterial Hypertension in Six Dogs. Vet. Pathol. 2006, 43, 510–522. [Google Scholar] [CrossRef]
- Disatian, S.; Ehrhart III, E.J.; Zimmerman, S.; Orton, E.C. Interstitial Cells from Dogs with Naturally Occurring Myxomatous Mitral Valve Disease Undergo Phenotype Transformation. J. Heart Valve Dis. 2008, 17, 402–412. [Google Scholar]
- Tuder, R.M.; Abman, S.H.; Braun, T.; Capron, F.; Stevens, T.; Thistlethwaite, P.A.; Haworth, S.G. Development and Pathology of Pulmonary Hypertension. J. Am. Coll. Cardiol. 2009, 54, S3–S9. [Google Scholar] [CrossRef]
- Säleby, J.; Bouzina, H.; Ahmed, S.; Lundgren, J.; Rådegran, G. Plasma Receptor Tyrosine Kinase RET in Pulmonary Arterial Hypertension Diagnosis and Differentiation. ERJ Open Res. 2019, 5, 00037–02019. [Google Scholar] [CrossRef] [PubMed]
- Borné, Y.; Gränsbo, K.; Nilsson, J.; Melander, O.; Orho-Melander, M.; Smith, J.G.; Engström, G. Vascular Endothelial Growth Factor D, Pulmonary Congestion, and Incidence of Heart Failure. J. Am. Coll. Cardiol. 2018, 71, 580–582. [Google Scholar] [CrossRef]
- Ahmed, S.; Ahmed, A.; Säleby, J.; Bouzina, H.; Lundgren, J.; Rådegran, G. Elevated Plasma Tyrosine Kinases VEGF-D and HER4 in Heart Failure Patients Decrease after Heart Transplantation in Association with Improved Haemodynamics. Heart Vessel. 2020, 35, 786–799. [Google Scholar] [CrossRef]
- Ray, L.; Mathieu, M.; Jespers, P.; Hadad, I.; Mahmoudabady, M.; Pensis, A.; Motte, S.; Peters, I.R.; Naeije, R.; McEntee, K. Early Increase in Pulmonary Vascular Reactivity with Overexpression of Endothelin-1 and Vascular Endothelial Growth Factor in Canine Experimental Heart Failure. Exp. Physiol. 2008, 93, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Oleynikov, D.A.; Yi, M. Pre-and Postcapillary Pulmonary Hypertension in Dogs: Circulating Biomarkers. Open Vet. J. 2022, 12, 469–480. [Google Scholar] [CrossRef]
- Sakarin, S.; Rungsipipat, A.; Roytrakul, S.; Jaresitthikunchai, J.; Phaonakrop, N.; Charoenlappanit, S.; Thaisakun, S.; Surachetpong, S.D. Proteomic Analysis of Pulmonary Arteries and Lung Tissues from Dogs Affected with Pulmonary Hypertension Secondary to Degenerative Mitral Valve Disease. PLoS ONE 2024, 19, e0296068. [Google Scholar] [CrossRef]
- Nakamura, T.; Kataoka, K.; Fukuda, M.; Nako, H.; Tokutomi, Y.; Dong, Y.-F.; Ichijo, H.; Ogawa, H.; Kim-Mitsuyama, S. Critical Role of Apoptosis Signal-Regulating Kinase 1 in Aldosterone/Salt-Induced Cardiac Inflammation and Fibrosis. Hypertension 2009, 54, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, L.D.; Darling, N.J.; Nicolaou, S.; Gori, I.; Squair, D.R.; Cohen, P.; Hill, C.S.; Sapkota, G.P. Salt-Inducible Kinases (SIKs) Regulate TGFβ-Mediated Transcriptional and Apoptotic Responses. Cell Death Dis. 2020, 11, 49. [Google Scholar] [CrossRef] [PubMed]
- Franco, O.E.; Tyson, D.R.; Konvinse, K.C.; Udyavar, A.R.; Estrada, L.; Quaranta, V.; Crawford, S.E.; Hayward, S.W. Altered TGF-α/β Signaling Drives Cooperation between Breast Cancer Cell Populations. FASEB J. 2016, 30, 3441. [Google Scholar] [CrossRef]
- Takahashi, H.; Shibuya, M. The Vascular Endothelial Growth Factor (VEGF)/VEGF Receptor System and Its Role under Physiological and Pathological Conditions. Clin. Sci. 2005, 109, 227–241. [Google Scholar] [CrossRef]
- Riengvirodkij, N.; Roytrakul, S.; Jaresitthikunchai, J.; Phaonakrop, N.; Charoenlappanich, S.; Sakcamduang, W. Peptide Barcodes in Dogs Affected by Mitral Valve Disease with and without Pulmonary Hypertension Using MALDI-TOF MS and LC-MS/MS. PLoS ONE 2021, 16, e0255611. [Google Scholar] [CrossRef]
- Kapanci, Y.; Burgan, S.; Pietra, G.G.; Conne, B.; Gabbiani, G. Modulation of Actin Isoform Expression in Alveolar Myofibroblasts (Contractile Interstitial Cells) during Pulmonary Hypertension. Am. J. Pathol. 1990, 136, 881–889. [Google Scholar]
- Glaus, T.M.; Soldati, G.; Ehrensperger, F.; Maurer, R. Clinical and Pathological Characterisation of Primary Pulmonary Hypertension in a Dog. Vet. Rec. 2004, 154, 786–789. [Google Scholar] [CrossRef]
- Chen, D.-D.; Dong, Y.-G.; Yuan, H.; Chen, A.F. Endothelin 1 Activation of Endothelin A Receptor/NADPH Oxidase Pathway and Diminished Antioxidants Critically Contribute to Endothelial Progenitor Cell Reduction and Dysfunction in Salt-Sensitive Hypertension. Hypertension 2012, 59, 1037–1043. [Google Scholar] [CrossRef]
- Kazimierczyk, R.; Kamiński, K. The Role of Platelets in the Development and Progression of Pulmonary Arterial Hypertension. Adv. Med. Sci. 2018, 63, 312–316. [Google Scholar] [CrossRef]
- Schoenichen, C.; Bode, C.; Duerschmied, D. Role of Platelet Serotonin in Innate Immune Cell Recruitment. Front. Biosci. Landmark 2019, 24, 514–526. [Google Scholar]
- Roumier, A.; Béchade, C.; Maroteaux, L. Serotonin and the Immune System. In Serotonin; Elsevier: Amsterdam, The Netherlands, 2019; pp. 181–196. [Google Scholar]
- Herr, N.; Bode, C.; Duerschmied, D. The Effects of Serotonin in Immune Cells. Front. Cardiovasc. Med. 2017, 4, 48. [Google Scholar] [CrossRef] [PubMed]
- Fudim, M.; Salah, H.M.; Sathananthan, J.; Bernier, M.; Pabon-Ramos, W.; Schwartz, R.S.; Rodés-Cabau, J.; Côté, F.; Khalifa, A.; Virani, S.A.; et al. Lymphatic Dysregulation in Patients with Heart Failure. J. Am. Coll. Cardiol. 2021, 78, 66–76. [Google Scholar] [CrossRef]
- Salah, H.M.; Biegus, J.; Ponikowski, P.P.; Fudim, M. Role of Lymphatics in Heart Failure. J. Soc. Cardiovasc. Angiogr. Interv. 2023, 2, 101204. [Google Scholar] [CrossRef]
- Shibuya, M.; Claesson-Welsh, L. Signal Transduction by VEGF Receptors in Regulation of Angiogenesis and Lymphangiogenesis. Exp. Cell Res. 2006, 312, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. The Crucial Role of Vascular Permeability Factor/Vascular Endothelial Growth Factor in Angiogenesis: A Historical Review. Br. J. Haematol. 2005, 128, 303–309. [Google Scholar] [CrossRef]
- Grünewald, F.S.; Prota, A.E.; Giese, A.; Ballmer-Hofer, K. Structure–Function Analysis of VEGF Receptor Activation and the Role of Coreceptors in Angiogenic Signaling. BBA-Proteins Proteom. 2010, 1804, 567–580. [Google Scholar] [CrossRef]
- Tammela, T.; Enholm, B.; Alitalo, K.; Paavonen, K. The Biology of Vascular Endothelial Growth Factors. Cardiovasc. Res. 2005, 65, 550–563. [Google Scholar] [CrossRef]
- Testa, U.; Pannitteri, G.; Condorelli, G.L. Vascular Endothelial Growth Factors in Cardiovascular Medicine. J. Cardiovasc. Med. 2008, 9, 1190–1221. [Google Scholar] [CrossRef]
- Jaffey, J.A.; Wiggen, K.; Leach, S.B.; Masseau, I.; Girens, R.E.; Reinero, C.R. Pulmonary Hypertension Secondary to Respiratory Disease and/or Hypoxia in Dogs: Clinical Features, Diagnostic Testing and Survival. Vet. J. 2019, 251, 105347. [Google Scholar] [CrossRef]
- Kellihan, H.B.; Waller, K.R.; Pinkos, A.; Steinberg, H.; Bates, M.L. Acute Resolution of Pulmonary Alveolar Infiltrates in 10 Dogs with Pulmonary Hypertension Treated with Sildenafil Citrate: 2005–2014. J. Vet. Cardiol. 2015, 17, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Townsley, M.I.; Fu, Z.; Mathieu-Costello, O.; West, J.B. Pulmonary Microvascular Permeability: Responses to High Vascular Pressure after Induction of Pacing-Induced Heart Failure in Dogs. Circ. Res. 1995, 77, 317–325. [Google Scholar] [CrossRef]
- Drake, R.E.; Doursout, M.F. Pulmonary Edema and Elevated Left Atrial Pressure: Four Hours and Beyond. Physiology 2002, 17, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Guglielmini, C.; Poser, H.; Dalla Pria, A.; Drigo, M.; Mazzotta, E.; Berlanda, M.; Luciani, A. Red Blood Cell Distribution Width in Dogs with Chronic Degenerative Valvular Disease. J. Am. Vet. Med. Assoc. 2013, 243, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Camacho, R.R.; Carvalho, E.R.; Pereira, E.Z.; Gava, F.N.; Camacho, A.A.; Sousa, M.G. Inflammatory Profile in Dogs with Myxomatous Mitral Valve Disease. Arch. Vet. Sci. 2017, 22. [Google Scholar] [CrossRef]
- Brložnik, M.; Pečjak, A.; Svete, A.N.; Petrič, A.D. Selected Hematological, Biochemical, and Echocardiographic Variables as Predictors of Survival in Canine Patients with Myxomatous Mitral Valve Disease and Congestive Heart Failure. J. Vet. Cardiol. 2023, 46, 18–29. [Google Scholar] [CrossRef]
- Guglielmini, C.; Valentini, C.M.; Contiero, B.; Valente, C.; Poser, H. Red Cell Distribution Width Has a Negative Prognostic Role in Dogs with Myxomatous Mitral Valve Disease. Animals 2021, 11, 778. [Google Scholar] [CrossRef]
- Mazzotta, E.; Guglielmini, C.; Menciotti, G.; Contiero, B.; Baron Toaldo, M.; Berlanda, M.; Poser, H. Red Blood Cell Distribution Width, Hematology, and Serum Biochemistry in Dogs with Echocardiographically Estimated Precapillary and Postcapillary Pulmonary Arterial Hypertension. J. Vet. Intern. Med. 2016, 30, 1806–1815. [Google Scholar] [CrossRef] [PubMed]
- Swann, J.W.; Sudunagunta, S.; Covey, H.L.; English, K.; Hendricks, A.; Connolly, D.J. Evaluation of Red Cell Distribution Width in Dogs with Pulmonary Hypertension. J. Vet. Cardiol. 2014, 16, 227–235. [Google Scholar] [CrossRef]
- Tangmahakul, N.; Orton, E.C.; Surachetpong, S.D. Investigation of Red Blood Cell and Platelet Indices in Adult Dogs Suffered from Myxomatous Mitral Valve Disease with and without Pulmonary Hypertension. Front. Vet. Sci. 2023, 10, 1234768. [Google Scholar] [CrossRef]
- Lerman, A.; Hildebrand, F.L., Jr.; Margulies, K.B.; O’murchu, B.; Perrella, M.A.; Heublein, D.M.; Schwab, T.R.; Burnett, J.C., Jr. Endothelin: A New Cardiovascular Regulatory Peptide. In Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 1990; Volume 65, pp. 1441–1455. [Google Scholar]
- Tamirisa, P.; Frishman, W.H.; Kumar, A. Endothelin and Endothelin Antagonism: Roles in Cardiovascular Health and Disease. Am. Heart J. 1995, 130, 601–610. [Google Scholar] [CrossRef]
- Shah, R. Endothelins in health and disease. Eur. J. Intern. Med. 2007, 18, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Nakamichi, K.; Ihara, M.; Kobayashi, M.; Saeki, T.; Ishikawa, K.; Yano, M. Different Distribution of Endothelin Receptor Subtypes in Pulmonary Tissues Revealed by the Novel Selective Ligands BQ-123 and [Ala1, 3, 11, 15] ET-1. Biochem. Biophys. Res. Commun. 1992, 182, 144–150. [Google Scholar] [CrossRef]
- Dupuis, J.; Goresky, C.A.; Fournier, A. Pulmonary Clearance of Circulating Endothelin-1 in Dogs in Vivo: Exclusive Role of ETBreceptors. J. Appl. Physiol. 1996, 81, 1510–1515. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Yung, G.L.; Marsh, J.J.; Konopka, R.G.; Pedersen, C.A.; Chiles, P.G.; Morris, T.A.; Channick, R.N. Endothelin Mediates Pulmonary Vascular Remodelling in a Canine Model of Chronic Embolic Pulmonary Hypertension. Eur. Respir. J. 2000, 15, 640–648. [Google Scholar] [CrossRef]
- Edmonston, D.L.; Parikh, K.S.; Rajagopal, S.; Shaw, L.K.; Abraham, D.; Grabner, A.; Sparks, M.A.; Wolf, M. Pulmonary Hypertension Subtypes and Mortality in CKD. Am. J. Kidney Dis. 2020, 75, 713–724. [Google Scholar] [CrossRef]
- Braz, J.B.; Beluque, T.; Ampuero, R.A.N.; Canola, R.A.M.; Batalhão, M.E.; Cárnio, E.C.; Camacho, A.A. Plasma Nitric Oxide in Dogs with Pulmonary Hypertension Secondary or Not to Left-Sided Heart Disease. Arq. Bras. Med. Vet. Zootec. 2023, 75, 161–173. [Google Scholar] [CrossRef]
- Tangmahakul, N.; Makoom, P.; Surachetpong, S.D. Assessment of Platelet and Plasma Serotonin in Canine Pulmonary Hypertension Secondary to Degenerative Mitral Valve Disease. Front. Vet. Sci. 2021, 8, 669. [Google Scholar] [CrossRef]
- Tangmahakul, N.; Sakarin, S.; Techangamsuwan, S.; Rungsipipat, A.; Surachetpong, S.D. Investigation of Genes and Proteins Expression Associating Serotonin Signaling Pathway in Lung and Pulmonary Artery Tissues of Dogs with Pulmonary Hypertension Secondary to Degenerative Mitral Valve Disease: The Preliminary Study. Vet. Sci. 2022, 9, 530. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.J.; Landzberg, M.J.; Anderson, T.J.; Charbonneau, F.; Creager, M.A.; Ganz, P.; Selwyn, A.P. Role of Nitric Oxide in the Local Regulation of Pulmonary Vascular Resistance in Humans. Circulation 1996, 93, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Nawarskas, J.; Henrie, A. Clinical Utility of Tadalafil in the Treatment of Pulmonary Arterial Hypertension: An Evidence-Based Review. Core Evid. 2015, 10, 99–109. [Google Scholar] [CrossRef] [PubMed]
- De Laforcade, A.M.; Freeman, L.M.; Rush, J.E. Serum Nitrate and Nitrite in Dogs with Spontaneous Cardiac Disease. Vet. Intern. Med. 2003, 17, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.F.; Quinn, R.L.; Rahilly, L.J. Biomarkers for Differentiation of Causes of Respiratory Distress in Dogs and Cats: Part 1–Cardiac Diseases and Pulmonary Hypertension. J. Vet. Emerg. Crit. Care 2015, 25, 311–329. [Google Scholar] [CrossRef] [PubMed]
- Reimann, M.J.; Häggström, J.; Mortensen, A.; Lykkesfeldt, J.; Møller, J.E.; Falk, T.; Olsen, L.H. Biopterin Status in Dogs with Myxomatous Mitral Valve Disease Is Associated with Disease Severity and Cardiovascular Risk Factors. Vet. Intern. Med. 2014, 28, 1520–1526. [Google Scholar] [CrossRef]
- Pedersen, H.D.; Schutt, T.; Søndergaard, R.; Qvortrup, K.; Olsen, L.H.; Kristensen, A.T. Decreased Plasma Concentration of Nitric Oxide Metabolites in Dogs with Untreated Mitral Regurgitation. Vet. Intern. Med. 2003, 17, 178–184. [Google Scholar] [CrossRef]
- Ghalayini, I.F. Nitric Oxide–Cyclic GMP Pathway with Some Emphasis on Cavernosal Contractility. Int. J. Impot. Res. 2004, 16, 459–469. [Google Scholar] [CrossRef]
- Ueda, Y.; Johnson, L.R.; Ontiveros, E.S.; Visser, L.C.; Gunther-Harrington, C.T.; Stern, J.A. Effect of a Phosphodiesterase-5A (PDE5A) Gene Polymorphism on Response to Sildenafil Therapy in Canine Pulmonary Hypertension. Sci. Rep. 2019, 9, 6899. [Google Scholar] [CrossRef]
- Hampl, V.; Herget, J. Role of Nitric Oxide in the Pathogenesis of Chronic Pulmonary Hypertension. Physiol. Rev. 2000, 80, 1337–1372. [Google Scholar] [CrossRef]
- Santhanam, A.V.R.; Viswanathan, S.; Dikshit, M. Activation of Protein Kinase B/Akt and Endothelial Nitric Oxide Synthase Mediates Agmatine-Induced Endothelium-Dependent Relaxation. Eur. J. Pharmacol. 2007, 572, 189–196. [Google Scholar] [CrossRef]
- Sasaki, A.; Doi, S.; Mizutani, S.; Azuma, H. Roles of Accumulated Endogenous Nitric Oxide Synthase Inhibitors, Enhanced Arginase Activity, and Attenuated Nitric Oxide Synthase Activity in Endothelial Cells for Pulmonary Hypertension in Rats. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2007, 292, L1480–L1487. [Google Scholar] [CrossRef]
- Mülsch, A.; Bauersachs, J.; Schäfer, A.; Stasch, J.-P.; Kast, R.; Busse, R. Effect of YC-1, an NO-Independent, Superoxide-Sensitive Stimulator of Soluble Guanylyl Cyclase, on Smooth Muscle Responsiveness to Nitrovasodilators. Br. J. Pharmacol. 1997, 120, 681–689. [Google Scholar] [CrossRef]
- Reyes-García, J.; Carbajal-García, A.; Di Mise, A.; Zheng, Y.-M.; Wang, X.; Wang, Y.-X. Important Functions and Molecular Mechanisms of Mitochondrial Redox Signaling in Pulmonary Hypertension. Antioxidants 2022, 11, 473. [Google Scholar] [CrossRef] [PubMed]
- Milstien, S.; Katusic, Z. Oxidation of Tetrahydrobiopterin by Peroxynitrite: Implications for Vascular Endothelial Function. Biochem. Biophys. Res. Commun. 1999, 263, 681–684. [Google Scholar] [CrossRef]
- Ontkean, M.; Gay, R.; Greenberg, B. Diminished Endothelium-Derived Relaxing Factor Activity in an Experimental Model of Chronic Heart Failure. Circ. Res. 1991, 69, 1088–1096. [Google Scholar] [CrossRef]
- Tjostheim, S.S.; Kellihan, H.B.; Grint, K.A.; Stepien, R.L. Effect of Sildenafil and Pimobendan on Intracardiac Heartworm Infections in Four Dogs. J. Vet. Cardiol. 2019, 23, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Jaffey, J.A.; Leach, S.B.; Kong, L.R.; Wiggen, K.E.; Bender, S.B.; Reinero, C.R. Clinical Efficacy of Tadalafil Compared to Sildenafil in Treatment of Moderate to Severe Canine Pulmonary Hypertension: A Pilot Study. J. Vet. Cardiol. 2019, 24, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Monassier, L.; Maroteaux, L. Serotonin and Cardiovascular Diseases. Serotonin 2019, 203–238. [Google Scholar] [CrossRef]
- Hanthazi, A.; Jespers, P.; Vegh, G.; Degroot, G.-N.; Springael, J.-Y.; Lybaert, P.; Dewachter, L.; Mc Entee, K. Chemerin Influences Endothelin-and Serotonin-Induced Pulmonary Artery Vasoconstriction in Rats. Life Sci. 2019, 231, 116580. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Fu, Z.; Hu, J.; Huang, C.; Paudel, O.; Cai, S.; Liedtke, W.; Sham, J.S. TRPV4 Channel Contributes to Serotonin-Induced Pulmonary Vasoconstriction and the Enhanced Vascular Reactivity in Chronic Hypoxic Pulmonary Hypertension. Am. J. Physiol.-Cell Physiol. 2013, 305, C704–C715. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Xu, P.; Tan, Y.; You, C.; Zhang, Y.; Jiang, Y.; Xu, H.E. Structural Basis for Recognition of Anti-Migraine Drug Lasmiditan by the Serotonin Receptor 5-HT1F–G Protein Complex. Cell Res. 2021, 31, 1036–1038. [Google Scholar] [CrossRef]
- Barman, S.A.; Pauly, J.R.; Isales, C.M. Canine Pulmonary Vasoreactivity to Serotonin: Role of Protein Kinase C and Tyrosine Kinase. Am. J. Physiol. 1997, 272, H740–H747. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, A.; Kroneberg, G.; Stoepel, K.; Stotzer, H. The Effects of Acute and Chronic Administration of Sympathomimetic Substances on the Systemic and Pulmonary Circulation. Proc. Eur. Soc. Drug Toxicol. 1970, 11, 110–117. [Google Scholar]
- Barman, S.A.; Isales, C.M. Fenfluramine Potentiates Canine Pulmonary Vasoreactivity to Endothelin-1. Pulm. Pharmacol. Ther. 1998, 11, 183–187. [Google Scholar] [CrossRef]
- Wang, H.Y.; Friedman, E. Central 5-Hydroxytryptamine Receptor-Linked Protein Kinase C Translocation: A Functional Postsynaptic Signal Transduction System. Mol. Pharmacol. 1990, 37, 75–79. [Google Scholar] [PubMed]
- Arndt, J.W.; Reynolds, C.A.; Singletary, G.E.; Connolly, J.M.; Levy, R.J.; Oyama, M.A. Serum Serotonin Concentrations in Dogs with Degenerative Mitral Valve Disease. J. Vet. Intern. Med. 2009, 23, 1208–1213. [Google Scholar] [CrossRef]
- Ni, W.; Watts, S.W. 5-Hydroxytryptamine in the Cardiovascular System: Focus on the Serotonin Transporter (SERT). Clin. Exp. Pharmacol. Physiol. 2006, 33, 575–583. [Google Scholar] [CrossRef]
- Torres, G.E.; Gainetdinov, R.R.; Caron, M.G. Plasma Membrane Monoamine Transporters: Structure, Regulation and Function. Nat. Rev. Neurosci. 2003, 4, 13–25. [Google Scholar] [CrossRef]
- Mangklabruks, T.; Surachetpong, S.D. Plasma and Platelet Serotonin Concentrations in Healthy Dogs and Dogs with Myxomatous Mitral Valve Disease. J. Vet. Cardiol. 2014, 16, 155–162. [Google Scholar] [CrossRef]
- Carneiro, A.M.D.; Cook, E.H.; Murphy, D.L.; Blakely, R.D. Interactions between Integrin αIIbβ3 and the Serotonin Transporter Regulate Serotonin Transport and Platelet Aggregation in Mice and Humans. J. Clin. Investig. 2008, 118, 1544–1552. [Google Scholar] [CrossRef]
- Mercado, C.P.; Kilic, F. Molecular Mechanisms of SERT in Platelets: Regulation of Plasma Serotonin Levels. Mol. Interv. 2010, 10, 231. [Google Scholar] [CrossRef]
- Lim, S.-J.; Lee, S.-H.; Song, K.-H. Serum Serotonin Concentration in Small Breed Dogs with Degenerative Mitral Valve Disease. J. Biomed. Translat. Res. 2015, 16, 177–181. [Google Scholar] [CrossRef]
- White, K.; Loughlin, L.; Maqbool, Z.; Nilsen, M.; McClure, J.; Dempsie, Y.; Baker, A.H.; MacLean, M.R. Serotonin Transporter, Sex, and Hypoxia: Microarray Analysis in the Pulmonary Arteries of Mice Identifies Genes with Relevance to Human PAH. Physiol. Genom. 2011, 43, 417–437. [Google Scholar] [CrossRef]
- Kim, K.-T.; Park, H.-M.; Hyun, C.; Seo, K.-W.; Song, K.-H. Serum Serotonin Concentration in Lean and Obese Dogs with Myxomatous Mitral Valve Disease. Korean J. Vet. Res. 2017, 56, 205–208. [Google Scholar] [CrossRef]
- Reimann, M.J.; Fredholm, M.; Cremer, S.E.; Christiansen, L.B.; Meurs, K.M.; Møller, J.E.; Häggström, J.; Lykkesfeldt, J.; Olsen, L.H. Polymorphisms in the Serotonin Transporter Gene and Circulating Concentrations of Neurotransmitters in Cavalier King Charles Spaniels with Myxomatous Mitral Valve Disease. J. Vet. Intern. Med. 2021, 35, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Cremer, S.E.; Kristensen, A.T.; Reimann, M.J.; Eriksen, N.B.; Petersen, S.F.; Marschner, C.B.; Tarnow, I.; Oyama, M.A.; Olsen, L.H. Plasma and Serum Serotonin Concentrations and Surface-Bound Platelet Serotonin Expression in Cavalier King Charles Spaniels with Myxomatous Mitral Valve Disease. Am. J. Vet. 2015, 76, 520–531. [Google Scholar] [CrossRef]
- Ljungvall, I.; Höglund, K.; Lilliehöök, I.; Oyama, M.A.; Tidholm, A.; Tvedten, H.; Häggström, J. Serum Serotonin Concentration Is Associated with Severity of Myxomatous Mitral Valve Disease in Dogs. J. Vet. Intern. Med. 2013, 27, 1105–1112. [Google Scholar] [CrossRef]
- Höglund, K.; Häggström, J.; Hanås, S.; Merveille, A.-C.; Gouni, V.; Wiberg, M.; Willesen, J.L.; Mc Entee, K.; Sørensen, L.M.; Tiret, L. Interbreed Variation in Serum Serotonin (5-Hydroxytryptamine) Concentration in Healthy Dogs. J. Vet. Cardiol. 2018, 20, 244–253. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grzeczka, A.; Pasławska, U.; Graczyk, S.; Antosik, P.; Zawadzki, M.; Pasławski, R. Pulmonary Hypertension Secondary to Myxomatous Mitral Valve Disease in Dogs: Current Insights into the Histological Manifestation and Its Determining Factors. Appl. Sci. 2024, 14, 2577. https://doi.org/10.3390/app14062577
Grzeczka A, Pasławska U, Graczyk S, Antosik P, Zawadzki M, Pasławski R. Pulmonary Hypertension Secondary to Myxomatous Mitral Valve Disease in Dogs: Current Insights into the Histological Manifestation and Its Determining Factors. Applied Sciences. 2024; 14(6):2577. https://doi.org/10.3390/app14062577
Chicago/Turabian StyleGrzeczka, Arkadiusz, Urszula Pasławska, Szymon Graczyk, Paulina Antosik, Marcin Zawadzki, and Robert Pasławski. 2024. "Pulmonary Hypertension Secondary to Myxomatous Mitral Valve Disease in Dogs: Current Insights into the Histological Manifestation and Its Determining Factors" Applied Sciences 14, no. 6: 2577. https://doi.org/10.3390/app14062577