Featured Application
The work aims to provide a comprehensive overview with pictorial examples of the neuroimaging characteristics of genetic GRIN-related epilepsies.
Abstract
N-methyl-D-aspartate receptors (NMDARs) are ionotropic glutamate channels that play a pivotal role in brain development and the regulation of learning and memory processes. De novo pathogenic variants in four genes encoding NMDA receptor subunits (GRIN1, GRIN2A, GRIN2B, and GRIN2D) have been implicated in a broad spectrum of neurodevelopmental disorders, including developmental delay, intellectual disability, autism spectrum disorders, epilepsy, and movement disorders. Mutations in the GRIN1 and GRIN2B genes, which encode the GluN1 and GluN2B subunits, respectively, are strongly associated with malformations of cortical development, including diffuse dysgyria, bilateral polymicrogyria, hippocampal dysplasia, corpus callosum hypoplasia, and other findings such as ventricular enlargement and basal ganglia abnormalities. Conversely, GRIN2A mutations are associated with heterogeneous and less specific neuroimaging patterns. We reviewed the existing literature on the neuroradiological features associated with GRIN gene mutations, also providing pictorial representations from our patient cohort. The analysis revealed a more consistent association of malformations of cortical development with GRIN1 and GRIN2B variants, likely reflecting the critical role of these genes in neuronal migration and proper development of cortical structures. In comparison, GRIN2A mutations are associated with milder brain abnormalities. An integrated assessment of neuroimaging patterns and GRIN gene variants provides valuable insights for differential diagnosis and supports targeted genetic screening in patients presenting with epileptic encephalopathy, global developmental delay, and autism spectrum disorders.
1. Introduction
N-methyl-D-aspartate receptors (NMDARs) are ionotropic glutamate receptors that are essential for excitatory neurotransmission, synaptic plasticity, and long-term potentiation, key elements in learning, memory, and brain development [1,2]. These receptors are tetrameric hetero-multimers comprising two GluN1 subunits and two variable GluN2A-D or GluN3A-B subunits, which are expressed differently during embryonic development of the central nervous system and in postnatal life [3]. They are characterized by high calcium permeability, requiring two ligands—glutamate and glycine—to be activated and a voltage-dependent Mg2+ block [1,3]. Most NMDA receptors expressed by glutamatergic excitatory synapses are represented by the GluN1/2A/2B tri-heteromer [3,4]. In addition to their expression at the synaptic membrane, NMDARs are also expressed extra synaptically, where they facilitate the synchronization of neuronal discharges and facilitate the propagation of action potentials in the soma and in the neuronal dendrites [3].
To understand the fundamental role of these receptors in the nervous system, it is worth noting that NMDARs are present early in the embryonic development of the central nervous system (CNS), even before the formation of synapses. They facilitate the influx of calcium into cells for signal transduction, gene transcription, and consequently the migration and maturation of cortical progenitor cells [5,6].
The analysis of the expression and distribution of the different NMDAR subunits in both animal models and the human brain shows different patterns for each subunit [3,7,8].
The GluN1 subunit, encoded by the GRIN1 gene, is ubiquitously expressed in the nervous system from the eighth week of human gestation and throughout postnatal life [3], being an obligate subunit for receptor formation. During prenatal life, GRIN1 expression in the fetal cerebral cortex increases with age [9]. A similar pattern is observed in the expression of GRIN1 gene transcripts in the white matter [7].
The GluN2 subunits, on the other hand, are modulatory and differently expressed during brain development: GluN2B and GluN2D are expressed at higher levels during embryonic development, while GluN2A and GluN2C are highly expressed after birth [3]. Murine models have shown that the expression of GluN2A begins a few days after birth throughout the CNS, while GluN2B is expressed ubiquitously in the early stages of postnatal brain development and then undergoes a progressive reduction in expression with a predominant localization in the forebrain [3]. Studies on the expression of the GluN2 subunits in the developing human brain show a gradual increase in the expression of the GluN2A subunit in late gestation, accompanied by a reduction in the expression of the GluN2B subunit and the corresponding gene [9].
In particular, the expression of these proteins is abundant in areas with high proliferation rates in the fetal brain (proliferative ventricular zone/subventricular zone compartments, the transient migratory zones, the intermediate zone and the subplate, and in the neuron-rich cortical plate), particularly in radial glial cells and multipotent cortical progenitors. Analysis of mid-gestation human cerebral cortex revealed expression of all NMDAR subunits, concurrent with active neurodevelopment. Transcript levels of GRIN1 and GRIN2A rise with fetal age, while GRIN2B decreases. Subunit protein quantities consistently reflect these gene expression dynamics. Hence, GRIN subunits orchestrate cortical development through spatiotemporally regulated expression, with GRIN1/GRIN2A promoting later-stage neuronal maturation and synaptogenesis, while GRIN2B drives early neurodevelopmental processes [9].
The GluN2A-GluN2B switch appears to occur not only in the expression levels of these subunits in the developing brain, but also in the distribution of these subunits in neurons. Specifically, GluN2B-rich NMDARs were found at the synaptic level during the formation of the synapses themselves [10]. On the other hand, GluN2A-rich NMDARs are more abundant in the synaptic membrane at a later stage, when synaptic activity is predominantly regulated by environmental stimuli [9,10]; at the same time, GluN2B-rich receptors translocate to the extrasynaptic level. Furthermore, the increase in the GluN2A/GluN2B ratio is also confirmed throughout post-natal life and during adulthood. The role of NMDARs in CNS development has also been confirmed by in vitro studies using receptor antagonists (e.g., kynurenic acid) on cell cultures derived from human fetal brain tissue [11]: these studies show that inhibition of NMDA receptors leads to a reduction in the proliferation and survival of radial glial cells with a consequent reduction in cortical progenitors, neurons, and interneurons; on the other hand, a greater differentiation towards the astrocytic line was observed, associated with an increase in the inflammatory response. Studies on in vivo animal models show that inhibition of NMDA glutamatergic receptors leads to alterations in the formation of cerebral cortex layers, cortical dysplasia, and heterotopias of neuronal groups [12].
In humans, pathogenic de novo variants in four genes encoding NMDAR subunits (GRIN1, GRIN2A, GRIN2B, and GRIN2D) have been identified in patients with neurodevelopmental disorders, including developmental delay (DD), intellectual disability (ID), autism spectrum disorder (ASD), epilepsy, and movement disorders [13]. Additionally, these clinical presentations may be variably associated with malformations of cortical development (MCDs).
Patients with de novo heterozygous mutations of the GRIN1 gene show profound DD from the neonatal period onwards, with subsequent severe/profound ID, significant motor disability with axial and sometimes limb hypotonia, followed by the development of pyramidal signs and eventually spastic tetraparesis. Most remain nonverbal and nonambulatory. Many patients also have hyperkinetic movement disorders associated with oculomotor abnormalities (oculogyric crises) or stereotypical movements. Epilepsy is often severe, with earlier onset compared to GRIN2A/GRIN2B (infantile or early childhood), with different types of seizures (e.g., infantile spasms, tonic and atonic seizures, hyperkinetic seizures, tonic-clonic seizures, possibly status epilepticus). Severe autistic traits and sleep disturbances are also common, often overshadowed by profound ID and motor impairment. Feeding difficulties and CVI have also been described [14,15].
The clinical presentation of patients with GRIN2B mutations is characterized by DD that progresses over time into moderate/severe ID, autistic features with language impairment, and seizures. Developmental regression has been described in a subset. Some patients achieve ambulation and limited speech. The epilepsy phenotype may be heterogeneous in terms of age of onset, seizure semiology, and EEG characteristics. Seizures tend to occur in about 30–40% of patients during childhood, as epileptic spasms, focal onset, or tonic-clonic seizures in various combinations. Drug resistance is reported in half of the cases [13]. Ataxia, spasticity, and hypotonia have been commonly reported. Less frequent findings include microcephaly, cortical visual impairment (CVI), and hyperkinetic movement disorders. The overall neurological phenotype may be troublesome, with hypotonia and tube-feeding as possible complications. These features can partially overlap with those encountered in other GRIN-related encephalopathies, especially those due to mutations in GRIN1 and in GRIN2D [14,15,16,17,18].
Mutations of GRIN2D, a modulatory subunit expressed strongly in the developing brainstem, thalamus, and spinal cord, with lower cortical expression, can also promote a severe neurological phenotype. Patients have moderate/severe ID, and epilepsy is very common and early-onset (infancy/childhood) with spasms, generalized tonic-clonic, myoclonic, or focal seizures. Hyperkinetic movement disorders, autistic traits, hypotonia, and feeding difficulties are also described [18].
The phenotypic spectrum reported in GRIN2A-related disorders is different. Patients can exhibit DD/ID and epilepsy with variable severity, but some affected individuals have normal cognitive abilities. GRIN2A-related epilepsy is typically associated with epilepsy-aphasia syndromes spanning over a continuous electro-clinical phenotype, from self-limiting epilepsy with centrotemporal spikes to epileptic and/or developmental encephalopathy with spike-wave activation during sleep/Landau-Kleffner syndrome. Seizures usually arise during pre-school or school age. Language and speech disorders are often encountered, including dysarthria, dyspraxia, aphasia, and nonverbal phenotypes. Ataxia, movement disorders (dystonia or choreiform movements), hypotonia, and other neuropsychiatric problems (behavioral disorders, attention deficit/hyperactivity disorder, autism spectrum disorder, schizophrenia, anxiety disorders, and mood disorders) are also described [19].
To summarize, pathogenic variants in GRIN genes cause a broad spectrum of neurodevelopmental disorders that often include DD/ID, autism, movement disorders, and epilepsy. Phenotypes vary by subunit: GRIN1 and GRIN2D mutations usually lead to profound developmental impairment, severe epilepsy, hypotonia, and feeding difficulties; GRIN2B variants yield a more heterogeneous phenotype, ranging from intellectual disability with autism and language delay (with or without epilepsy) to severe developmental and epileptic encephalopathies. Mutations of GRIN2A often present with epilepsy–aphasia syndromes and variable cognitive outcomes. These differences highlight the critical developmental role of NMDARs and the need for precision approaches to diagnosis and treatment in GRIN-related disorders.
2. Materials and Methods
We undertook systematic searches of PubMed, EMBASE, and Google Scholar to identify relevant studies published between 1 January 2000 and 1 July 2025. The search strategy combined the terms “GRIN” OR “GRIN gene” OR “NMDA receptor” with “neurodevelopmental disorder” OR “epilepsy” OR “seizure” AND “MRI” OR “neuroimaging”. Articles were eligible if they reported cases or cohorts of individuals with GRIN-related neurodevelopmental disorders and/or epilepsy and included brain MRI data. Studies without neuroimaging information were excluded. Titles and abstracts were screened, followed by full-text review. Reference lists of reviews and case series were examined to identify additional eligible reports, and duplicate cases were removed. Eligible publications included peer-reviewed case reports, case series, cohort studies, and reviews. From the final set, data on neuroimaging features—including cortical dysplasia, polymicrogyria, hippocampal abnormalities, basal ganglia alterations, and other malformations—were assessed for qualitative synthesis. We also reviewed the MRIs of patients with GRIN-related mutations performed in our Institute (IRCCS Stella Maris Foundation, Pisa, Italy) to capture salient pictorial examples of neuroimaging findings. We obtained informed consent from the caregivers to collect the images for clinical purposes as well as for research and publication purposes.
We aimed to identify common neuroanatomical patterns associated with each GRIN gene mutation while highlighting differences in neuroimaging phenotypes across the gene variants.
3. Results
In published studies, we found 28 patients with pathogenic variants in GRIN1 or GRIN2B and MCDs. As discussed beforehand, the clinical phenotype of patients with GRIN1/GRIN2B may be similar: consistent DD/ID, motor and language impairment, movement disorders, autistic traits, epilepsy, and microcephaly (more common in patients with MCDs compared to those without MCDs). We found evidence of a common neuroradiological pattern between these two genes. The majority of patients have bilateral diffuse or bilateral perisylvian polymicrogyria (PMG). Bilateral fronto-parietal PMG has also been reported [13,20,21,22,23] (Table 1). Dysgyria, i.e., abnormal gyral pattern in terms of sulcal depth and orientation with normal cortical thickness, has also been commonly reported. Dysgyria was originally described in tubulinopathies and dystroglycanopathies [20], with bilateral distribution similar to GRIN-related disorders [24]. Distinguishing polymicrogyria and dysgyria might be cumbersome from a radiological standpoint. In previous works, individuals initially diagnosed with dysgyria due to GRIN1 variants have been reclassified as polymicrogyria after neuropathological examination [24]. Polymicrogyria/dysgyria are often associated with concomitant brain dysmorphism such as nodular heterotopias, dysplastic basal ganglia and hippocampi, hypoplasia of the corpus callosum or cerebellum. Different from tubulinopathies, no infratentorial abnormalities are reported [24]. Significant examples of major and minor GRIN-related findings are shown in the MRI images of our patients in Figure 1.

Table 1.
This table shows the percentage of MCDs or other anomalies found on the brain MRI of the patients described previously in the literature.
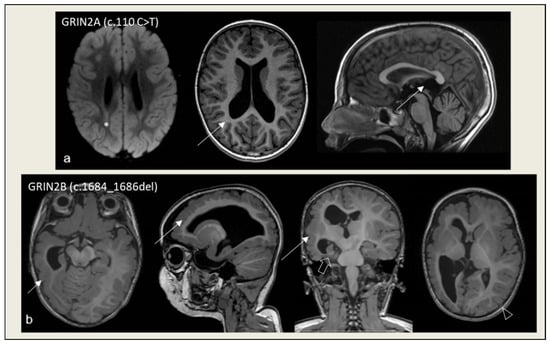
Figure 1.
Minor findings and overt MCDs in patients with GRIN-related disorders. (a) Mild ectasia of the posterior portions of the lateral ventricles and of the suprapineal recess of the third ventricle (arrows in T1W images); subtle hyperintensity in the posterior portion of the middle cells (asterisk in FLAIR image) at 3 years of age. (b) Right diffuse polymicrogyria (arrow in T1W images) with hemispheric hypoplasia and ventricular dysmorphism, right hippocampal malrotation/dysplasia (bold arrow in T1W image), limited polymicrogyric region on the left hemisphere (arrowhead in T1W image) at 1 year of age.
The functional effect of GRIN variants might be implicated in the development of MCDs. Most of the reported GRIN2B mutations in epileptic encephalopathies are missense and de novo. The main mechanism is a gain-of-function (GOF) [13], but loss-of-function (LOF) is also described [28]. Similarly, GRIN1 variants are missense and can lead to either GOF or LOF effects [21]. Functional studies have been performed in a few patients with MCDs, demonstrating that diffuse dysgyria is associated with GOF in both GRIN1 and GRIN2B. Gain-of-function mutations increase the sensitivity of the receptor to ligands (glutamate and glycine) by altering its gating; excessive activation of the NMDA receptor due to increased sensitivity to ligands can lead to excitotoxicity and early neuronal death, yielding an aberrant cortical morphology [21]. In general, animal models suggest that LOF mutations more rarely affect the cortical structure [31]; reduced NMDAR activity may compromise pro-survival signaling, leading to more gradual neuronal death and thus missing the key time window in which polymicrogyria/dysgyria can develop (5–22 weeks post-conception). The brain atrophy observed in some patients with LOF is likely due to both the direct effects of the mutations (excitotoxicity or loss of pro-survival signaling) and damage resulting from frequent seizures [21].
However, the genotype-phenotype correlation is only partial; GOF variants may also be detected in patients with normal neuroimaging [32]. We noted that mutations found in patients with MCDs affect specific protein domains, different from those found when neuroimaging is normal: S1-M1 linker, the Lurcher motif in the M3 region, the S2 domain of GluN1, M3, S2, and M4 domains of GluN2B [21,28]. An exception is the recurrent variant p.(Met818Thr) in GRIN2B, which has been reported in both a patient with and in a patient without MCDs; this may indicate that additional modulators (such as non-genetic events or genetic background) may contribute to the neuroimaging phenotype [13,24]. Likely, the specific variant type rather than the functional effect only can drive the neuroimaging pattern.
Unlike what has been observed in patients with mutations in GRIN1 and GRIN2B, we found no pattern of abnormal neuronal migration in GRIN2A-related cases. The GRIN2A gene is mostly expressed post-natally, with limited impact on neuronal migration. Knockdown of GluN1 and GluN2B subunits in rats grossly impairs the radial migration of neurons in the intermediate zone of the developing brain, while knockdown of GluN2A does not influence the correct trajectory of neurons towards the cortical plate [33].
Even though most patients with GRIN2A pathogenic variants have normal brain MRI (75.3%), generalized brain atrophy (11%), and other heterogeneous brain abnormalities have also been rarely reported (14.1%), including focal cortical dysplasia, hypoplasia/dysplasia of the corpus callosum, hippocampal abnormalities, neuronal heterotopia, subcortical lesions, and olfactory bulb hypoplasia [19]. The gene is remarkably less involved in neuronal migration compared to GRIN1/GRIN2B.
At the molecular level, missense mutations in GRIN2A located within the transmembrane and linker domains are associated with severe phenotypes and receptor GOF, whereas those in the extracellular ATD (amino-terminal domain) and LBD (ligand-binding domain) regions lead to LOF and milder phenotypes, often presenting with isolated language deficits and self-limited epilepsies [19]. Overall, these findings support the hypothesis of a distinctive and postnatal role for GluN2A, primarily in synaptic development, consistent with the absence of major cortical malformations in most GRIN2A-positive patients, but with a clinically significant impact on cognitive and language domains.
The study published for GRIN2D variants is too limited to draw firm conclusions. However, normal neuroimaging or minor non-specific abnormalities (cerebral/cerebellar atrophy, delayed myelination) have been reported so far [18].
4. Discussion
An in-depth analysis of mutations in GRIN1, GRIN2A, and GRIN2B reveals a complex and multifactorial landscape. Considering the structural and genetic etiopathogenesis underlying epileptic encephalopathies caused by variants of those genes, instrumental investigations (brain MRI and sleep-wake EEG) are essential in clinical practice to identify specific patterns that may guide targeted genetic screening. The intrinsic properties of mutations, which may result in either GOF or LOF effects, interact with environmental and epigenetic factors, as well as with differences in allelic expression, contributing to the considerable clinical heterogeneity observed even among individuals sharing the same pathogenic variant [29].
GRIN1-related disorders are characterized by striking seizure heterogeneity, with onset ranging from the neonatal period to late childhood and seizure types including infantile spasms, tonic, atonic, hypermotor, febrile, focal dyscognitive, and generalized seizures. Electroencephalographic findings are similarly diverse, ranging from hypsarrhythmia to multifocal epileptiform discharges, and while a subset of patients may respond to conventional antiseizure therapies, the majority develop drug-resistant epilepsy [15]. In contrast, GRIN2A mutations are most strongly associated with focal epilepsy syndromes, spanning a spectrum from benign rolandic epilepsy to atypical rolandic epilepsy and more severe epilepsy–aphasia disorders such as Landau–Kleffner syndrome and continuous spike–wave during slow-wave sleep; in some cases, infantile-onset developmental and epileptic encephalopathy has also been reported [19]. GRIN2B mutations, on the other hand, typically result in a mixed seizure phenotype with both focal and generalized seizures, often accompanied by epileptic spasms. Importantly, GRIN2B-related epilepsies are consistently associated with significant neurodevelopmental impairment, including intellectual disability and developmental delay, features that are less pronounced in GRIN2A and variable in GRIN1 [13].
From a neuroimaging standpoint, germinal variants in the GRIN gene can result in several types of MCDs. In general, these are characterized by abnormal cortical structure or the presence of heterotopic grey matter, possibly associated with micro/macrocephaly in children who may exhibit drug-resistant epilepsy, intellectual disability, cerebral palsy [20]. The proper diagnosis can only be obtained by a neuropathology exam; however, pathological tissue can only be obtained in patients who are surgically treated or after autopsy. Therefore, MCDs are constantly identified and classified based on neuroimaging features. Several classifications have been drafted over time, and the latest has summarized MCDs according to the presumed first-affected brain developmental stage: cell proliferation and apoptosis, cell migration, and post-migrational development. On behalf of the pan-European multidisciplinary network (COST Action Neuro-MIG), Severino et al. have produced a consensus paper of best practice guidelines for the radiological diagnosis of MCD that can assist in the judgment of an abnormal brain morphology [20].
Variants in the GRIN1 and GRIN2B genes have been associated with complex and early-onset MCDs such as polymicrogyria/dysgyria, hippocampal dysplasia, basal ganglia abnormalities, and corpus callosum anomalies, highlighting a direct impact on corticogenesis and neuronal migration [24]. Polymicrogyria/dysgyria are the most common. Polymicrogyria is characterized by an excessive number of abnormally small cerebral gyri, classified by Barkovich et al. as a malformation due to abnormal postmigrational development [34]. Polymicrogyria can vary in terms of distribution (focal–most commonly perisylvian-multifocal, generalized), laterality (unilateral, bilateral), and symmetry (symmetric/asymmetric). The etiology is heterogeneous and includes both genetic and acquired causes (e.g., in-utero infections) [35]. Dysgyria is a recently introduced term for a non-specific cortical malformation characterized by dysmorphic cortex, with normal thickness and abnormal gyral patterns (irregular sulcal depth/orientation, smooth cortical surface with radially oriented sulci, narrow gyri separated by abnormally deep/shallow sulci) [36]. Although dysgyria was not included in the latest classification of MCDs, it is commonly regarded as deriving from a post- or even late neuronal migration defect. In GRIN1/GRIN2B, polymicrogyria and dysgyria have been consistently described as bilateral, yet Avsenik et al. interestingly reported a couple of monozygotic twins in whom the same pathogenic variant in the GRIN2B gene (c.2453T > C) yielded a unilateral MCD in a patient and a bilateral MCD in the other [29]. In our experience, we can confirm a unilateral distribution is possible for GRIN2B, as observed in a patient with an intragenic deletion (Figure 1). The other reported MCDs are often ill-defined. The hypoplasia or agenesis of the corpus callosum is a very non-specific finding [37,38], even though commonly reported in various neurodevelopment disorders, both genetic or secondary to other conditions (premature birth, fetal alcohol syndrome) [39]. Hippocampal dysplasia is an umbrella term rather than a specific malformation, used to include hypoplasia/hyperplasia of the hippocampus as well as malrotations, and is often associated with other MCDs [40]. Aberrant basal ganglia, usually dysmorphic and enlarged, are encountered in a remarkable number of GRIN-mutated patients. In patients described by Brock et al. [24], harbouring GRIN-mutations and showing MCDs, hippocampal dysplasia was reported in 33% of GRIN1 and in 60% of GRIN2B, basal ganglia dysmorphisms in 45% of GRIN1 and 90% of GRIN2B, along with bilateral polymicrogyria/dysgyria. Of note, these malformations are rarely reported in patients with MCDs in general and may represent diagnostic clues for GRIN-related encephalopathies. Indeed, tubulinopathies may show hippocampal dysplasia/basal ganglia dysmorphism as well as bilateral dysgyria, but also exhibit abnormalities of the brainstem and cerebellum that are not reported in GRIN-mutated patients [41].
In contrast, in individuals carrying GRIN2A variants, structural brain malformations are rarer, more heterogeneous, and generally less pronounced. Only a minority of cases show abnormalities on imaging, supporting a more selective involvement of the GluN2A subunit in maturational processes rather than in early cortical morphogenesis [19]. This divergence aligns with the spatiotemporal expression profile of NMDA receptor subunits: GluN2B is highly expressed during prenatal development with a subsequent impairment in neuronal migration patterns associated with GRIN2B mutations, while GluN2A expression increases progressively in the postnatal period, coinciding with synaptic maturation [33]. Focal cortical dysplasia has been found in up to 14% of individuals with GRIN2A-related disorders. Less specific findings have also been occasionally described, such as hypoplasia of the corpus callosum, neuronal heterotopia, and hippocampal sclerosis [19]. In patients with normal brain MRI, minor abnormalities of cortical thickness have been found over the perisylvian regions, matching the classical speech-language involvement of GRIN2A [25].
Morphometric analyses conducted in patients with pathogenic variants in the GRIN2A gene have revealed an increased cortical thickness in the inferior frontal and superior temporal regions bilaterally, accompanied by a reduction in left hippocampal volume; the most pronounced alterations are observed in the left pars opercularis, a subregion of the posterior inferior frontal gyrus corresponding to Broca’s area. These perisylvian anomalies are consistent with the language impairment typically observed in individuals with GRIN2A mutations and resemble neuroanatomical patterns reported in other genetic neurodevelopmental disorders with overlapping phenotypic features. The neurodevelopmental mechanisms underlying increased cortical thickness in language-related regions remain poorly understood. In the typical trajectory of brain maturation, an age-related cortical thinning, particularly in frontal areas, is a well-documented process [42,43], while in several neurodevelopmental conditions, abnormal thickness of the cortex is reported [44,45]. It is hypothesized that GRIN2A mutations may impair NMDA receptor–dependent synaptic plasticity, particularly within the hippocampus [46], thereby affecting its maturation and its volume with a potentially secondary effect on language acquisition.
Therefore, the integrated analysis of neuroimaging findings in patients carrying GRIN mutations can provide valuable insights for differential diagnosis. The presence of a specific neuroimaging pattern may assist clinicians in directing targeted genetic investigations, particularly in individuals presenting with similar or overlapping phenotypical traits. To date, no direct cases of epilepsy surgery in GRIN mutation–positive patients have been reported. However, focal MCDs with robust electro-clinical correlation might theoretically be evaluated for epilepsy surgery, as in other structural genetic epilepsies. Of note, brain structural abnormalities are often associated with more severe developmental impairment and drug-resistant epilepsy, helping to set realistic expectations for families and clinicians. Moreover, a deeper understanding of the molecular mechanisms underlying MCDs may contribute to the development of novel targeted therapeutic strategies aimed at enhancing pro-survival signaling or mitigating excitotoxicity.
Studies have reported heterogeneous outcomes regarding the use of memantine in patients carrying GRIN2A and GRIN2B gain-of-function mutations [13,16]. Certain GRIN2A gene variants characterized by loss of Mg2+ block (e.g., p.Leu812Met) have demonstrated a more favorable response to memantine, whereas others with comparable biophysical properties (e.g., p.Met818Thr) have not shown significant clinical benefit. Patients carrying the GRIN2B mutation who received memantine in association with antiseizure medications (ASMs) showed improvements in awareness, behavior, and sleep quality, although no significant reduction in seizure frequency was observed. These variable responses are likely influenced by multiple factors, including the specific genetic variant, the underlying molecular mechanism, patient age, and individual pharmacokinetics (e.g., plasma or brain drug concentrations). Overall, the clinical efficacy of memantine remains uncertain; controlled studies are needed to clarify its therapeutic potential and guide individualized treatments. On top of memantine, other targeted treatments with NMDAR antagonists (memantine, ketamine, dextromethorphan for gain-of-function variants) or co-agonists (glycine, D-serine for loss-of-function variants) may be considered [47]. Neuroimaging adds an important layer to this therapeutic decision-making. When MRI reveals structural abnormalities, seizures may be predominantly driven by the malformed cortex, and patients may require complex polytherapy or consideration of surgical/palliative options, with a limited role for NMDAR modulators. Conversely, a normal MRI increases the likelihood that epileptogenesis is primarily related to receptor dysfunction, thereby strengthening the rationale for precision pharmacological approaches such as memantine in gain-of-function variants. Indeed, longitudinal MRI can also be valuable for tracking progressive cortical changes (e.g., atrophy), providing additional context for adapting treatment strategies over time. Thus, neuroimaging, alongside genetic and electrophysiological data, is clinically relevant not only for diagnosis but also for stratifying patients toward targeted therapy versus conventional multimodal treatment pathways.
5. Limitations
This review is qualitative in nature; therefore, no quantitative assessments or standardized quality indices were applied to evaluate the methodological rigor of the included studies. Another limitation concerns the heterogeneity of the included sources, which consist of case reports, case series, and review articles—study types that inherently differ in terms of their level of evidence. This variability may affect the generalizability and consistency of the findings. Additionally, in many of the included articles, information regarding patients’ brain MRIs was not supported by actual images, which prevented a direct evaluation of the reported findings. Moreover, several studies did not provide key technical details, such as the magnetic field strength used or the specific MRI sequences performed. These limitations result in neuroimaging findings of varying quality, making them difficult to compare across studies and potentially affecting the consistency and reliability of the overall analysis. Furthermore, the small sample size makes it difficult to generalize the collected data.
6. Conclusions
Growing evidence of a mixed etiology (genetic and structural) in forms linked to mutations in these genes supports the need for an integrated diagnostic approach that systematically includes brain MRI in patients with GRIN gene pathogenic variants, as the clinical phenotype may be modulated by the coexistence of cortical malformations.
A multidisciplinary approach integrating genetic data, structural neuroimaging, and sleep/wake EEG appears essential for early diagnosis, accurate phenotypic classification, and targeted therapeutic management of disorders associated with NMDAR receptor dysfunction. Through this approach, it is possible to develop personalized strategies and improve clinical outcomes in patients with these complex encephalopathies.
Author Contributions
Conceptualization, E.B.; methodology, E.B.; data curation, E.B. and R.P.; writing—original draft preparation, M.C., A.A. and I.M.; writing—review and editing, E.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was partially funded by the Italian Ministry of Health via the RC-grant and 5 × 1000 voluntary contributions.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Pediatric Ethics Committee of Tuscany Region, Italy (GEF-EpiPed study, approval code is 131/2024, date of approval 9/7/2024).
Informed Consent Statement
Written informed consent has been obtained from the patients and caregivers to depict their neuroimaging studies.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Korinek, M.; Candelas Serra, M.; Abdel Rahman, F.; Dobrovolski, M.; Kuchtiak, V.; Abramova, V.; Fili, K.; Tomovic, E.; Hrcka Krausova, B.; Krusek, J.; et al. Disease-Associated Variants in GRIN1, GRIN2A and GRIN2B Genes: Insights into NMDA Receptor Structure, Function, and Pathophysiology. Physiol. Res. 2024, 73, S413–S434. [Google Scholar] [CrossRef]
- Liu, L.; Wong, T.P.; Pozza, M.F.; Lingenhoehl, K.; Wang, Y.; Sheng, M.; Auberson, Y.P.; Wang, Y.T. Role of NMDA Receptor Subtypes in Governing the Direction of Hippocampal Synaptic Plasticity. Science 2004, 304, 1021–1024. [Google Scholar] [CrossRef]
- Hansen, K.B.; Wollmuth, L.P.; Bowie, D.; Furukawa, H.; Menniti, F.S.; Sobolevsky, A.I.; Swanson, G.T.; Swanger, S.A.; Greger, I.H.; Nakagawa, T.; et al. Structure, Function, and Pharmacology of Glutamate Receptor Ion Channels. Pharmacol. Rev. 2021, 73, 298–487. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wang, Y.; Yasuda, R.P.; Dunah, A.W.; Wolfe, B.B. The Majority of N-Methyl-D-Aspartate Receptor Complexes in Adult Rat Cerebral Cortex Contain at Least Three Different Subunits (NR1/NR2A/NR2B). Mol. Pharmacol. 1997, 51, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Jansson, L.C.; Åkerman, K.E. The Role of Glutamate and Its Receptors in the Proliferation, Migration, Differentiation and Survival of Neural Progenitor Cells. J. Neural Transm. 2014, 121, 819–836. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, N.C. Electrical Activity in Early Neuronal Development. Nature 2006, 444, 707–712. [Google Scholar] [CrossRef]
- Jantzie, L.L.; Talos, D.M.; Jackson, M.C.; Park, H.-K.; Graham, D.A.; Lechpammer, M.; Folkerth, R.D.; Volpe, J.J.; Jensen, F.E. Developmental Expression of N-Methyl-d-Aspartate (NMDA) Receptor Subunits in Human White and Gray Matter: Potential Mechanism of Increased Vulnerability in the Immature Brain. Cereb. Cortex 2015, 25, 482–495. [Google Scholar] [CrossRef]
- Henson, M.A.; Roberts, A.C.; Salimi, K.; Vadlamudi, S.; Hamer, R.M.; Gilmore, J.H.; Jarskog, L.F.; Philpot, B.D. Developmental Regulation of the NMDA Receptor Subunits, NR3A and NR1, in Human Prefrontal Cortex. Cereb. Cortex 2008, 18, 2560–2573. [Google Scholar] [CrossRef]
- Bagasrawala, I.; Memi, F.V.; Radonjić, N.; Zecevic, N. N-Methyl d-Aspartate Receptor Expression Patterns in the Human Fetal Cerebral Cortex. Cereb. Cortex 2017, 27, 5041–5053. [Google Scholar] [CrossRef]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA Receptor Subunit Diversity: Impact on Receptor Properties, Synaptic Plasticity and Disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Bagasrawala, I.; Zecevic, N.; Radonjić, N.V. N-Methyl D-Aspartate Receptor Antagonist Kynurenic Acid Affects Human Cortical Development. Front. Neurosci. 2016, 10, 435. [Google Scholar] [CrossRef]
- Reiprich, P.; Kilb, W.; Luhmann, H.J. Neonatal NMDA Receptor Blockade Disturbs Neuronal Migration in Rat Somatosensory Cortex In Vivo. Cereb. Cortex 2005, 15, 349–358. [Google Scholar] [CrossRef]
- Platzer, K.; Yuan, H.; Schütz, H.; Winschel, A.; Chen, W.; Hu, C.; Kusumoto, H.; Heyne, H.O.; Helbig, K.L.; Tang, S.; et al. GRIN2B Encephalopathy: Novel Findings on Phenotype, Variant Clustering, Functional Consequences and Treatment Aspects. J. Med. Genet. 2017, 54, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Lemke, J.R.; Geider, K.; Helbig, K.L.; Heyne, H.O.; Schütz, H.; Hentschel, J.; Courage, C.; Depienne, C.; Nava, C.; Heron, D.; et al. Delineating the GRIN1 Phenotypic Spectrum: A Distinct Genetic NMDA Receptor Encephalopathy. Neurology 2016, 86, 2171–2178. [Google Scholar] [CrossRef] [PubMed]
- Ohba, C.; Shiina, M.; Tohyama, J.; Haginoya, K.; Lerman-Sagie, T.; Okamoto, N.; Blumkin, L.; Lev, D.; Mukaida, S.; Nozaki, F.; et al. GRIN1 Mutations Cause Encephalopathy with Infantile-Onset Epilepsy, and Hyperkinetic and Stereotyped Movement Disorders. Epilepsia 2015, 56, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.M.; Yuan, H.; Marsh, E.D.; Fuentes-Fajardo, K.; Adams, D.R.; Markello, T.; Golas, G.; Simeonov, D.R.; Holloman, C.; Tankovic, A.; et al. GRIN2A Mutation and Early-Onset Epileptic Encephalopathy: Personalized Therapy with Memantine. Ann. Clin. Transl. Neurol. 2014, 1, 190–198. [Google Scholar] [CrossRef]
- Endele, S.; Rosenberger, G.; Geider, K.; Popp, B.; Tamer, C.; Stefanova, I.; Milh, M.; Kortüm, F.; Fritsch, A.; Pientka, F.K.; et al. Mutations in GRIN2A and GRIN2B Encoding Regulatory Subunits of NMDA Receptors Cause Variable Neurodevelopmental Phenotypes. Nat. Genet. 2010, 42, 1021–1026. [Google Scholar] [CrossRef]
- Platzer, K.; Krey, I.; Lemke, J.R. GRIN2D-Related Developmental and Epileptic Encephalopathy. In GeneReviews® [Internet]; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2022; pp. 1993–2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK582335/ (accessed on 1 April 2025).
- Strehlow, V.; Heyne, H.O.; Vlaskamp, D.R.M.; Marwick, K.F.M.; Rudolf, G.; de Bellescize, J.; Biskup, S.; Brilstra, E.H.; Brouwer, O.F.; Callenbach, P.M.C.; et al. GRIN2A-Related Disorders: Genotype and Functional Consequence Predict Phenotype. Brain 2019, 142, 80–92. [Google Scholar] [CrossRef]
- Severino, M.; Geraldo, A.F.; Utz, N.; Tortora, D.; Pogledic, I.; Klonowski, W.; Triulzi, F.; Arrigoni, F.; Mankad, K.; Leventer, R.J.; et al. Definitions and Classification of Malformations of Cortical Development: Practical Guidelines. Brain 2020, 143, 2874–2894. [Google Scholar] [CrossRef]
- Fry, A.E.; Fawcett, K.A.; Zelnik, N.; Yuan, H.; Thompson, B.A.N.; Shemer-Meiri, L.; Cushion, T.D.; Mugalaasi, H.; Sims, D.; Stoodley, N.; et al. De Novo Mutations in GRIN1 Cause Extensive Bilateral Polymicrogyria. Brain 2018, 141, 698–712. [Google Scholar] [CrossRef]
- Nishimura, N.; Kumaki, T.; Murakami, H.; Enomoto, Y.; Katsumata, K.; Toyoshima, K.; Kurosawa, K. Arthrogryposis Multiplex Congenita with Polymicrogyria and Infantile Encephalopathy Caused by a Novel GRIN1 Variant. Hum. Genome Var. 2020, 7, 29. [Google Scholar] [CrossRef]
- Platzer, K.; Lemke, J.R. GRIN2B-Related Neurodevelopmental Disorder. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Brock, S.; Laquerriere, A.; Marguet, F.; Myers, S.J.; Hongjie, Y.; Baralle, D.; Vanderhasselt, T.; Stouffs, K.; Keymolen, K.; Kim, S.; et al. Overlapping Cortical Malformations in Patients with Pathogenic Variants in GRIN1 and GRIN2B. J. Med. Genet. 2023, 60, 183–192. [Google Scholar] [CrossRef]
- Thompson-Lake, D.G.Y.; Liegeois, F.J.; Braden, R.O.; Jackson, G.D.; Turner, S.J.; Morison, L.; Hildebrand, M.; Scheffer, I.E.; Morgan, A.T. Perisylvian and Hippocampal Anomalies in Individuals with Pathogenic GRIN2A Variants. Neurol. Genet. 2024, 10, e200129. [Google Scholar] [CrossRef] [PubMed]
- Sculier, C.; Tilmant, A.-S.; De Tiège, X.; Giurgea, S.; Paquier, P.; Rudolf, G.; Lesca, G.; Van Bogaert, P. Acquired Epileptic Opercular Syndrome Related to a Heterozygous Deleterious Substitution in GRIN2A. Epileptic Disord. 2017, 19, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Mangano, G.D.; Riva, A.; Fontana, A.; Salpietro, V.; Mangano, G.R.; Nobile, G.; Orsini, A.; Iacomino, M.; Battini, R.; Astrea, G.; et al. De Novo GRIN2A Variants Associated with Epilepsy and Autism and Literature Review. Epilepsy Behav. 2022, 129, 108604. [Google Scholar] [CrossRef] [PubMed]
- Smigiel, R.; Kostrzewa, G.; Kosinska, J.; Pollak, A.; Stawinski, P.; Szmida, E.; Bloch, M.; Szymanska, K.; Karpinski, P.; Sasiadek, M.M.; et al. Further Evidence for GRIN2B Mutation as the Cause of Severe Epileptic Encephalopathy. Am. J. Med. Genet. A 2016, 170, 3265–3270. [Google Scholar] [CrossRef]
- Avsenik, J.; Benedik, M.P.; Rogač, M.; Biswas, A.; Sudhakar, S.; D’Arco, F.; Löbel, U.; Mankad, K. Divergent Presentation of GRIN2B Neurodevelopmental Disorder in Monozygotic Twins: Case Report with Unique Imaging Phenotypes. Neuropediatrics 2025, 56, 269–273. [Google Scholar] [CrossRef]
- Sharawat, I.K.; Yadav, J.; Saini, L. Novel GRIN2B Mutation: A Rare Cause of Severe Epileptic Encephalopathy. Neurol. India 2019, 67, 562–563. [Google Scholar] [CrossRef]
- Messersmith, E.K.; Feller, M.B.; Zhang, H.; Shatz, C.J. Migration of Neocortical Neurons in the Absence of Functional NMDA Receptors. Mol. Cell Neurosci. 1997, 9, 347–357. [Google Scholar] [CrossRef]
- Crino, P.B. Polymicrogyria and GRIN1 Mutations: Altered Connections, Altered Excitability. Brain 2018, 141, 622–623. [Google Scholar] [CrossRef]
- Jiang, H.; Jiang, W.; Zou, J.; Wang, B.; Yu, M.; Pan, Y.; Lin, Y.; Mao, Y.; Wang, Y. The GluN2B Subunit of N-Methy-D-Asparate Receptor Regulates the Radial Migration of Cortical Neurons In Vivo. Brain Res. 2015, 1610, 20–32. [Google Scholar] [CrossRef]
- Barkovich, A.J.; Kuzniecky, R.I.; Jackson, G.D.; Guerrini, R.; Dobyns, W.B. A Developmental and Genetic Classification for Malformations of Cortical Development. Neurology 2005, 65, 1873–1887. [Google Scholar] [CrossRef]
- Raybaud, C.; Widjaja, E. Development and Dysgenesis of the Cerebral Cortex: Malformations of Cortical Development. Neuroimaging Clin. N. Am. 2011, 21, 483–543. [Google Scholar] [CrossRef]
- Mutch, C.A.; Poduri, A.; Sahin, M.; Barry, B.; Walsh, C.A.; Barkovich, A.J. Disorders of Microtubule Function in Neurons: Imaging Correlates. AJNR Am. J. Neuroradiol. 2016, 37, 528–535. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, V.; Boccherini, C.; Manganaro, L.; Panici, P.B.; Cellitti, R.; Vena, F.; Pajno, C.; Corno, S.; Brunelli, R.; Giancotti, A. Hypoplasia of the Corpus Callosum: A Single Center Experience and a Concise Literature Review. Fetal Pediatr. Pathol. 2021, 40, 626–637. [Google Scholar] [CrossRef] [PubMed]
- Hofman, J.; Hutny, M.; Sztuba, K.; Paprocka, J. Corpus Callosum Agenesis: An Insight into the Etiology and Spectrum of Symptoms. Brain Sci. 2020, 10, 625. [Google Scholar] [CrossRef] [PubMed]
- Paul, L.K. Developmental Malformation of the Corpus Callosum: A Review of Typical Callosal Development and Examples of Developmental Disorders with Callosal Involvement. J. Neurodev. Disord. 2011, 3, 3–27. [Google Scholar] [CrossRef]
- Montenegro, M.A.; Kinay, D.; Cendes, F.; Bernasconi, A.; Bernasconi, N.; Coan, A.C.; Li, L.M.; Guerreiro, M.M.; Guerreiro, C.A.M.; Lopes-Cendes, I.; et al. Patterns of Hippocampal Abnormalities in Malformations of Cortical Development. J. Neurol. Neurosurg. Psychiatry 2006, 77, 367–371. [Google Scholar] [CrossRef][Green Version]
- Bahi-Buisson, N.; Poirier, K.; Fourniol, F.; Saillour, Y.; Valence, S.; Lebrun, N.; Hully, M.; Bianco, C.F.; Boddaert, N.; Elie, C.; et al. The Wide Spectrum of Tubulinopathies: What Are the Key Features for the Diagnosis? Brain 2014, 137, 1676–1700. [Google Scholar] [CrossRef]
- McGinnis, S.M.; Brickhouse, M.; Pascual, B.; Dickerson, B.C. Age-Related Changes in the Thickness of Cortical Zones in Humans. Brain Topogr. 2011, 24, 279–291. [Google Scholar] [CrossRef]
- Fjell, A.M.; Grydeland, H.; Krogsrud, S.K.; Amlien, I.; Rohani, D.A.; Ferschmann, L.; Storsve, A.B.; Tamnes, C.K.; Sala-Llonch, R.; Due-Tønnessen, P.; et al. Development and Aging of Cortical Thickness Correspond to Genetic Organization Patterns. Proc. Natl. Acad. Sci. USA 2015, 112, 15462–15467. [Google Scholar] [CrossRef]
- Shen, L.; Zhang, J.; Fan, S.; Ping, L.; Yu, H.; Xu, F.; Cheng, Y.; Xu, X.; Yang, C.; Zhou, C. Cortical Thickness Abnormalities in Autism Spectrum Disorder. Eur. Child Adolesc. Psychiatry 2024, 33, 65–77. [Google Scholar] [CrossRef]
- Kubota, M.; Miyata, J.; Yoshida, H.; Hirao, K.; Fujiwara, H.; Kawada, R.; Fujimoto, S.; Tanaka, Y.; Sasamoto, A.; Sawamoto, N.; et al. Age-Related Cortical Thinning in Schizophrenia. Schizophr. Res. 2011, 125, 21–29. [Google Scholar] [CrossRef]
- Kullmann, D.M.; Lamsa, K.P. Long-Term Synaptic Plasticity in Hippocampal Interneurons. Nat. Rev. Neurosci. 2007, 8, 687–699. [Google Scholar] [CrossRef]
- Samanta, D.; Bhatia, S.; Hunter, S.E.; Rao, C.K.; Xiong, K.; Karakas, C.; Reeders, P.C.; Erdemir, G.; Sattar, S.; Axeen, E.; et al. Current and Emerging Precision Therapies for Developmental and Epileptic Encephalopathies. Pediatr. Neurol. 2025, 168, 67–81. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).