
A Theoretical Study on the Influence of the Functional Group Electronic Effect on the Electron Mobility of Cross-Linked Polyethylene


Abstract
1. Introduction
2. Computational Methods
3. Results and Discussion
3.1. Theoretical Method Screening and Stationary Point Geometries
3.2. Frontier MOs and Electron Mobility
3.3. Wide Temperature Range Electron Mobility
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Orton, H. History of underground power cables. IEEE Electr. Insul. Mag. 2013, 29, 52–57. [Google Scholar] [CrossRef]
- Teyssedre, G.; Laurent, C. Advances in high-field insulating polymeric materials over the past 50 years. IEEE Electr. Insul. Mag. 2013, 29, 26–36. [Google Scholar] [CrossRef]
- He, J.L.; Dang, B.; Zhou, Y.; Hu, J. Reviews on Research Progress and Key Technology in Extruded Cables for HVDC Transmission. Gaodianya Jishu/High Volt. Eng. 2015, 41, 1417. [Google Scholar] [CrossRef]
- Adams, W.I. A historical perspective of HDPE piping material development. In Proceedings of the ASME pressure vessels and piping conference, Toronto, ON, Canada, 15–19 July 2012; Volume 1, pp. 325–333. [Google Scholar]
- Montanari, G.C.; Palmieri, F.; Motori, A.; Perego, G.; Serra, S.; Mazzanti, G. Space-charge trapping and conduction in LDPE, HDPE and XLPE. J. Phys. D Appl. Phys. 2001, 34, 2902–2911. [Google Scholar] [CrossRef]
- Fu, M.; Chen, G.; Dissado, L.A.; Fothergill, J.C. Influence of thermal treatment and residues on space charge accumulation in XLPE for DC power cable application. IEEE Trans. Dielectr. Electr. Insul. 2007, 14, 53–64. [Google Scholar] [CrossRef]
- Hussin, N.; Chen, G.; Lewis, T.J. Analysis of space charge formation in LDPE in the presence of crosslinking byproducts. IEEE Trans. Dielectr. Electr. Insul. 2012, 19, 126–133. [Google Scholar] [CrossRef]
- Cheng, F.C. Insulation thickness determination of polymeric power cables. IEEE Trans. Dielectr. Electr. Insul. 1994, 1, 624–629. [Google Scholar] [CrossRef]
- Dong, W.; Wang, X.; Tian, B.; Liu, Y.G.; Jiang, Z.X.; Li, Z.G.; Zhou, W. Use of Grafted Voltage Stabilizer to Enhance Dielectric Strength of Cross-Linked Polyethylene. Polymers 2019, 11, 176. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hu, J.; Dang, B.; He, J.L. Mechanism of highly improved electrical properties in polypropylene by chemical modification of grafting maleic anhydride. J. Phys. D Appl. Phys. 2016, 49, 415301. [Google Scholar] [CrossRef]
- Abou-Dakka, M.; Bulinski, A.; Bamji, S.S. Effect of additives on the performance of cross-linked polyethylene subjected to long term single and periodically reversed polarity DC voltage. IEEE Trans. Dielectr. Electr. Insul. 2013, 20, 654–663. [Google Scholar] [CrossRef]
- Neague, R.; Neagu, R.M. On the nature of the origin ofisothermal and non-isothermal current released fromdielectric materials. Thin Solid Films 2001, 384, 15–22. [Google Scholar] [CrossRef]
- Roy, S.L.; Teyssedre, G.; Laurent, C. Chargetransport and dissipative processes in insulating polymers: Experimentsand mode. IEEE Trans. Dielectr. Electr. Insul. 2005, 12, 644–654. [Google Scholar] [CrossRef]
- Roy, S.L.; Teyssedre, G.; Laurent, C.; Montanari, G.C.; Palmieri, F. Description of charge transport in polyethylene using afluid model with a constant mobility: Fitting model andexperiments. J. Phys. D Appl. Phys. 2006, 39, 1427–1436. [Google Scholar] [CrossRef]
- Boufayed, F.; Teyssedre, G.; Laurent, C.; Le-Roy, S.; Dissado, L.A.; Ségur, P.; Montanari, G.C. Models of bipolar charge transport in polyethylene. J. Appl. Phys. 2006, 100, 104–105. [Google Scholar] [CrossRef]
- Zhou, T.C.; Chen, G.; Liao, R.J.; Xu, Z. Charge trapping and detrapping inpolymeric materials. J. Appl. Phys. 2009, 106, 123707. [Google Scholar] [CrossRef]
- Zheng, Y.S.; Zhong, X.Y.; Miao, X.R.; He, J.L. Numerical simulations of dynamic space chargecharacteristics in low density Polyethylene under polarityreversal. High Volt. Eng. 2017, 43, 385–390. [Google Scholar] [CrossRef]
- Fu, Y.W.; Sun, W.F.; Wang, X. UV-Initiated Crosslinking Reaction Mechanism and Electrical Breakdown Performance of Crosslinked Polyethylene. Polymers 2020, 12, 420. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shen, J.; Yang, L.J.; Zhang, H.Z.; Zhao, Y.S. Influence of cooling medium on space charge property of LDPE. High Volt. Eng. 2010, 36, 2629–2633. [Google Scholar] [CrossRef]
- Yang, X.D.; Li, Q.K.; Shuai, Z.G. Theoretical modelling of carrier transports in molecular semiconductors: Molecular design of triphenylamine dimer systems. Nanotechnology 2007, 18, 424029. [Google Scholar] [CrossRef]
- Narbe, M.; Martin, H.G. Thirty years of density functional theory in computational chemistry: An overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Zhang, H.; Shang, Y.; Li, M.X.; Zhao, H.; Wang, X.; Han, B.Z. Theoretical study on the radical reaction mechanism in the cross-linking process of polyethylene. RSC Adv. 2015, 5, 90343–90353. [Google Scholar] [CrossRef]
- Zhu, R.; Li, Q.S.; Li, Z.S. Nitrogen substitution improves the mobility and stability of electron transport materials for inverted perovskite solar cells. Nanoscale 2018, 10, 17873–17883. [Google Scholar] [CrossRef]
- Coropceanu, V.; Cornil, J.; da Silva Filho, D.A.; Olivier, Y.; Silbey, R.; Brédas, J.L. Charge transport in organic semiconductors. Chem. Rev. 2007, 107, 926–952. [Google Scholar] [CrossRef]
- Deng, W.Q.; Goddard, W.A. Predictions of hole mobilities in oligoacene organic semiconductors from quantum mechanical calculations. J. Phys. Chem. B 2004, 108, 8614–8621. [Google Scholar] [CrossRef]
- Marcus, R.A. Electron transfer reactions in chemistry. Theory and experiment. Rev. Mod. Phys. 1993, 65, 599–610. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Watanabe, K.; Nakayama, T.; Mottl, J. Ionization potentials of some molecules. J. Quant. Spectry Radiat. Transf. 1962, 2, 369. [Google Scholar] [CrossRef]
- Liangyu, G.; Demin, T.; Bin, Q.; Liheng, W. Effect of ferrocene derivatives on the dielectric of polyethylene. In Proceedings of the IEEE International Conference on Properties & Applications of Dielectric Materials, Tokyo, Japan, 8–12 July 1991; Volume 2, pp. 796–799. [Google Scholar]
- Chen, X.R.; Yu, L.W.; Dai, C.; Paramane, A.; Liu, H.Y.; Wei, Z.J.; Tanaka, Y. Enhancement of Insulating Properties of Polyethylene Blends by Delocalization Type Voltage Stabilizers. IEEE Trans. Dielectr. Electr. Insul. 2019, 6, 2041–2049. [Google Scholar] [CrossRef]
- Hong, Z.L.; Chen, X.R.; Zhu, G.Y.; Awais, M.; Shi, Y.W.; Zhu, H.S.; Li, H.; Paramane, A. Inhibition of electrical trees degradation of crosslinked polyethylene at high temperatures by electron-buffering voltage stabilizers. J. Appl. Polym. Sci. 2022, 139, e52786. [Google Scholar] [CrossRef]
- Wei, Z.J.; Liu, H.Y.; Yu, L.W.; Xiao, S.W.; Hou, Y.X.; Chen, X.R. Delocalized aromatic molecules with matched electron-donating and electron-withdrawing groups enhancing insulating performance of polyethylene blends. J. Appl. Polym. Sci. 2020, 137, e49185. [Google Scholar] [CrossRef]
- Xia, J.F.; Zhang, Z.W.; Zheng, F.H.; Lei, Q.Q. Numerical simulation of charge packet behavior in low-density polyethylene based on Gunn effect-like model. Acta Phys. Sin. 2010, 59, 508–514. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Molecular Formula | Molecular Name | ab. | Molecular Formula | Molecular Name | ab. |
|---|---|---|---|---|---|
 | 3-Methylpentane | Pe |  | 3-Nitromethylpentane | Nmp |
 | 3-Aminobenzoic acid | Aba | 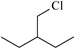 | 3-Benzylchloropentane | Bcp |
 | 3-dimethylaminopentane | Dap |  | 3-Ethyl-4-butylaniline | Eba |
 | 3-aminomethylpentane | Amp |  | 3-Ethyl-4-butyldimethylaminobenzene | Ebdb |
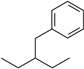 | 3-Ethylbutylbenzene | Ebb |  | 3-Ethyl-4-butylnitrobenzene | Ebnb |
 | 3-Ethyl-4-butyl-3-aminobenzoic acid | Ebaa |
| Method | EA(a) | IP(a) | Method | EA(a) | IP(a) |
|---|---|---|---|---|---|
| HF/6-311G(d,p) | −4.08 | 9.12 | M062X/6-311G(d,p) | −2.72 | 10.40 |
| HF/6-311+G(d,p) | −1.93 | 9.89 | M062X/6-311+G(d,p) | −1.34 | 10.40 |
| MP2/6-311G(d,p) | −3.13 | 10.53 | M062X/6-311G(3df,2p) | −2.42 | 10.37 |
| MP2/6-311+G(d,p) | −1.52 | 10.38 | wB97XD/6-311G* | −3.14 | 10.31 |
| PBE/6-311G(d,p) | −2.37 | 9.45 | wB97XD/6-311+G* | −1.72 | 10.30 |
| PBE/6-311+G(d,p) | −1.06 | 9.46 | wB97XD/6-311G(d,p) | −3.14 | 10.13 |
| B3LYP/6-311G(d,p) | −2.54 | 9.62 | wB97XD/6-311+G(d,p) | −1.72 | 10.26 |
| B3LYP/6-311+G(d,p) | −1.14 | 10.01 | wB97XD/6-311G(3df,2p) | −2.92 | 10.23 |
| B3LYP/6-311G(3df,2p) | −2.28 | 9.58 | Experimental data | -- | 10.08 [29] |
| Molecular Formula | λe | Eg | EA(a) | IP(a) | EA(v) | IP(v) |
|---|---|---|---|---|---|---|
| 0.16 | 13.77 | −3.14 | 10.13 (10.08) [29] | −3.21 | 11.04 |
| 0.65 | 4.47 (4.60) [32] | 0.16 | 7.78 | −0.16 | 8.10 |
| 0.19 | 10.94 | −2.91 | 7.27 | −3.01 | 8.04 |
| 0.22 | 11.56 | −2.82 | 8.27 | −2.93 | 9.11 |
| 0.52 | 10.36 | −1.51 | 8.47 | −1.75 | 8.67 |
| 1.55 | 10.57 | −0.02 | 10.36 | −0.67 | 10.90 |
| 7.05 | 12.97 | 0.29 | 10.11 | −2.88 | 10.64 |
| 0.73 | 8.42 | −0.20 | 7.37 | −0.57 | 7.81 |
| 0.66 | 8.92 | −1.47 | 6.74 | −1.80 | 6.95 |
| 0.51 | 9.22 | −1.55 | 7.10 | −1.80 | 7.47 |
| 0.69 | 8.95 | 0.72 | 9.99 | 0.36 | 10.70 |
| Molecular Formula | 233 K | 263 K | 273 K | 300 K | 313 K | 333 K | 363 K |
|---|---|---|---|---|---|---|---|
| 9.93 × 10−8 | 1.16 × 10−7 | 1.22 × 10−7 | 1.35 × 10−7 (1.7 × 10−7) [34] | 1.41 × 10−7 | 1.48 × 10−7 | 1.59 × 10−7 |
| 7.17 × 10−23 | 1.72 × 10−22 | 2.17 × 10−22 | 3.91 × 10−22 | 4.88 × 10−22 | 6.86 × 10−22 | 1.04 × 10−21 |
| 8.79 × 10−12 | 1.08 × 10−11 | 1.15 × 10−11 | 1.31 × 10−11 | 1.39 × 10−11 | 1.49 × 10−11 | 1.64 × 10−11 |
| 7.43 × 10−13 | 9.52 × 10−13 | 1.02 × 10−12 | 1.20 × 10−12 | 1.29 × 10−12 | 1.41 × 10−12 | 1.58 × 10−12 |
| 2.78 × 10−16 | 5.42 × 10−16 | 6.60 × 10−16 | 9.85 × 10−16 | 1.24 × 10−15 | 1.60 × 10−15 | 2.23 × 10−15 |
| 4.62 × 10−21 | 3.85 × 10−20 | 7.24 × 10−20 | 2.99 × 10−19 | 5.56 × 10−19 | 1.26 × 10−18 | 3.75 × 10−18 |
| 6.92 × 10−50 | 1.29 × 10−45 | 2.41 × 10−44 | 1.76 × 10−41 | 3.19 × 10−40 | 1.43 × 10−38 | 2.37 × 10−36 |
| 1.41 × 10−22 | 3.70 × 10−22 | 4.93 × 10−22 | 9.36 × 10−22 | 1.24 × 10−21 | 1.79 × 10−21 | 2.92 × 10−21 |
| 3.58 × 10−19 | 8.53 × 10−19 | 1.10 × 10−18 | 1.96 × 10−18 | 2.52 × 10−18 | 3.51 × 10−18 | 5.44 × 10−18 |
| 6.23 × 10−20 | 1.20 × 10−19 | 1.45 × 10−19 | 2.23 × 10−19 | 2.70 × 10−19 | 3.45 × 10−19 | 4.78 × 10−19 |
| 3.39 × 10−48 | 8.40 × 10−48 | 1.10 × 10−47 | 2.01 × 10−47 | 2.62 × 10−47 | 3.70 × 10−47 | 5.86 × 10−47 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, Y.; Zhang, H.; Du, X.; Shang, Y.; Wang, X.; Chen, Q.; Li, Z. A Theoretical Study on the Influence of the Functional Group Electronic Effect on the Electron Mobility of Cross-Linked Polyethylene. Appl. Sci. 2025, 15, 959. https://doi.org/10.3390/app15020959
Du Y, Zhang H, Du X, Shang Y, Wang X, Chen Q, Li Z. A Theoretical Study on the Influence of the Functional Group Electronic Effect on the Electron Mobility of Cross-Linked Polyethylene. Applied Sciences. 2025; 15(2):959. https://doi.org/10.3390/app15020959
Chicago/Turabian StyleDu, Yang, Hui Zhang, Xia Du, Yan Shang, Xuan Wang, Qingguo Chen, and Zesheng Li. 2025. "A Theoretical Study on the Influence of the Functional Group Electronic Effect on the Electron Mobility of Cross-Linked Polyethylene" Applied Sciences 15, no. 2: 959. https://doi.org/10.3390/app15020959
APA StyleDu, Y., Zhang, H., Du, X., Shang, Y., Wang, X., Chen, Q., & Li, Z. (2025). A Theoretical Study on the Influence of the Functional Group Electronic Effect on the Electron Mobility of Cross-Linked Polyethylene. Applied Sciences, 15(2), 959. https://doi.org/10.3390/app15020959