
A Gene Ontology-Based Pipeline for Selecting Significant Gene Subsets in Biomedical Applications


Abstract
1. Introduction and Literature Survey
2. Materials and Methods
2.1. Experimental Dataset
2.2. Gene Ontology-Based Pipeline for Identifying Statistically Significant Genes
- 1.
- Identification of Differentially Expressed Genes (DEGs). Statistical analysis is performed using the Kruskal–Wallis test, a non-parametric method suitable for comparing gene expression distributions across multiple independent groups. This test is particularly appropriate in the current study, as prior assessment revealed that the majority of gene expression profiles deviate from normality, often exhibiting skewed or multimodal distributions. Given the presence of 14 sample classes, the Kruskal–Wallis test enables the detection of genes with significant expression differences between groups. The output is a vector of p-values, one for each gene, indicating the probability that the observed differences would arise by chance under the null hypothesis.
- 2.
- Multiple Testing Correction Using the Benjamini–Hochberg (BH) Method [23]. Due to the large number of simultaneous hypothesis tests (tens of thousands of genes), the probability of false-positive results increases. The BH procedure addresses this issue by controlling the false discovery rate (FDR), which reflects the expected proportion of incorrect rejections among all rejected hypotheses.The method consists of several steps. First, the p-values obtained from the tests are sorted in ascending order. Then, each sorted is adjusted using the following formula:where m is the total number of hypotheses tested, while i is the index of the sorted p-value.To ensure monotonicity, if , then is set equal to . This step guarantees that the adjusted q-values do not decrease as the rank increases.Genes with adjusted q-values below a predefined threshold are considered statistically significant.
- 3.
- Threshold Initialization. To determine which genes should be selected for further analysis, a significance threshold is defined. In this study, the threshold was set to , which is a widely accepted value in statistical hypothesis testing. This value represents a 5% probability of committing a Type I error (false positive), balancing sensitivity and specificity in the identification of differentially expressed genes.The choice of this threshold was motivated by both statistical convention and practical considerations in high-dimensional biological data analysis. A more stringent threshold could exclude biologically relevant genes by increasing the false-negative rate, whereas a more lenient one might increase the risk of false discoveries. By applying in conjunction with the Benjamini–Hochberg correction for multiple testing, the procedure effectively controls the FDR while maintaining adequate sensitivity.
- 4.
- Gene Ranking. Genes are ranked in ascending order based on their adjusted q-values to prioritize those with the strongest statistical significance.
- 5.
- GO Enrichment Analysis. To identify biologically meaningful functional groups among the differentially expressed genes, enrichment analysis is conducted using the topGO package https://bioconductor.org/packages/release/bioc/html/topGO.html (accessed on 29 October 2024) [24]. The analysis is performed separately for each of the three principal Gene Ontology (GO) categories: Biological Process (BP), Molecular Function (MF), and Cellular Component (CC).The analysis uses a gene universe consisting of all genes tested in the differential expression analysis, with significance determined by adjusted q-values. The selection function identifies genes with q-values below the threshold . To ensure functional relevance and statistical robustness, only GO terms with a minimum of 10 annotated genes are considered.The enrichment of GO terms is evaluated using two complementary statistical tests. First, Fisher’s exact test is applied to detect the overrepresentation of significant genes within each GO term. Second, the Kolmogorov–Smirnov (KS) test is used to assess distributional shifts in gene-level scores across GO terms.GO terms are retained for downstream analysis only if they are found to be statistically significant by both tests. Gene annotations are mapped using the org.Hs.eg.db database, and all gene identifiers are standardized to the Entrez format.
- 6.
- Extraction of Significant Genes Associated with Enriched GO Terms. Genes that are linked to significantly enriched GO terms—validated by both Fisher’s exact test and the Kolmogorov–Smirnov test—are selected for further analysis. The corresponding Entrez IDs of these genes are extracted and aggregated across all three GO categories (BP, MF, and CC). The resulting gene list is composed of unique identifiers that appear in at least one of the enriched GO categories.
- 7.
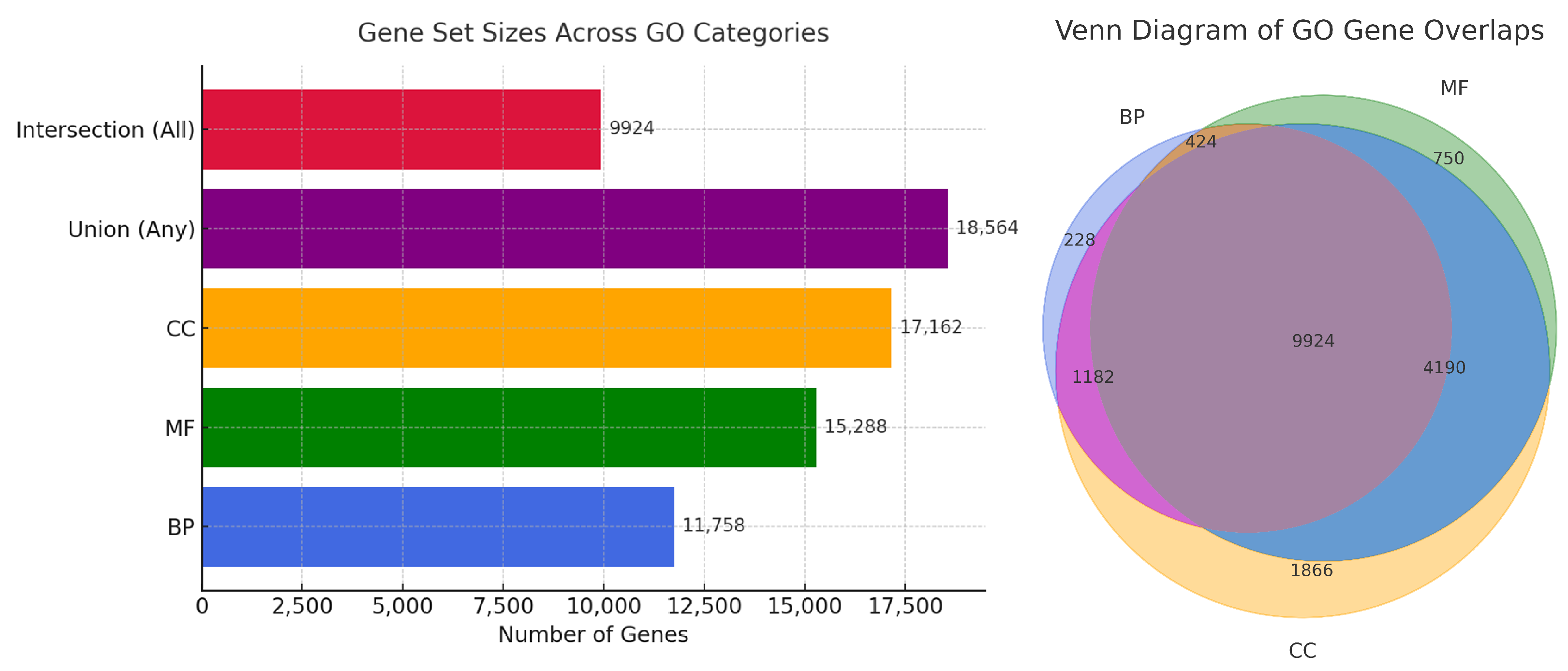
- Construction of the Final Dataset. Based on the normalized expression profiles of the selected significant genes, are distinct datasets were constructed to support downstream analysis. The first three datasets were category-specific: the BP-specific dataset contained genes identified as significant within the Biological Process (BP) category; the MF-specific dataset included genes significant in the Molecular Function (MF) category; and the CC-specific dataset comprised genes significant in the Cellular Component (CC) category.In addition, two integrative datasets were created. The intersection dataset included only those genes that were found to be significant in all three GO categories simultaneously, representing the intersection of BP, MF, and CC. The union dataset, by contrast, contained unique genes that were significant in at least one of the GO categories.Each of these datasets preserves the normalized expression values of the selected genes across all 6344 samples and serves as the foundation for the subsequent classification and evaluation procedures.
- 8.
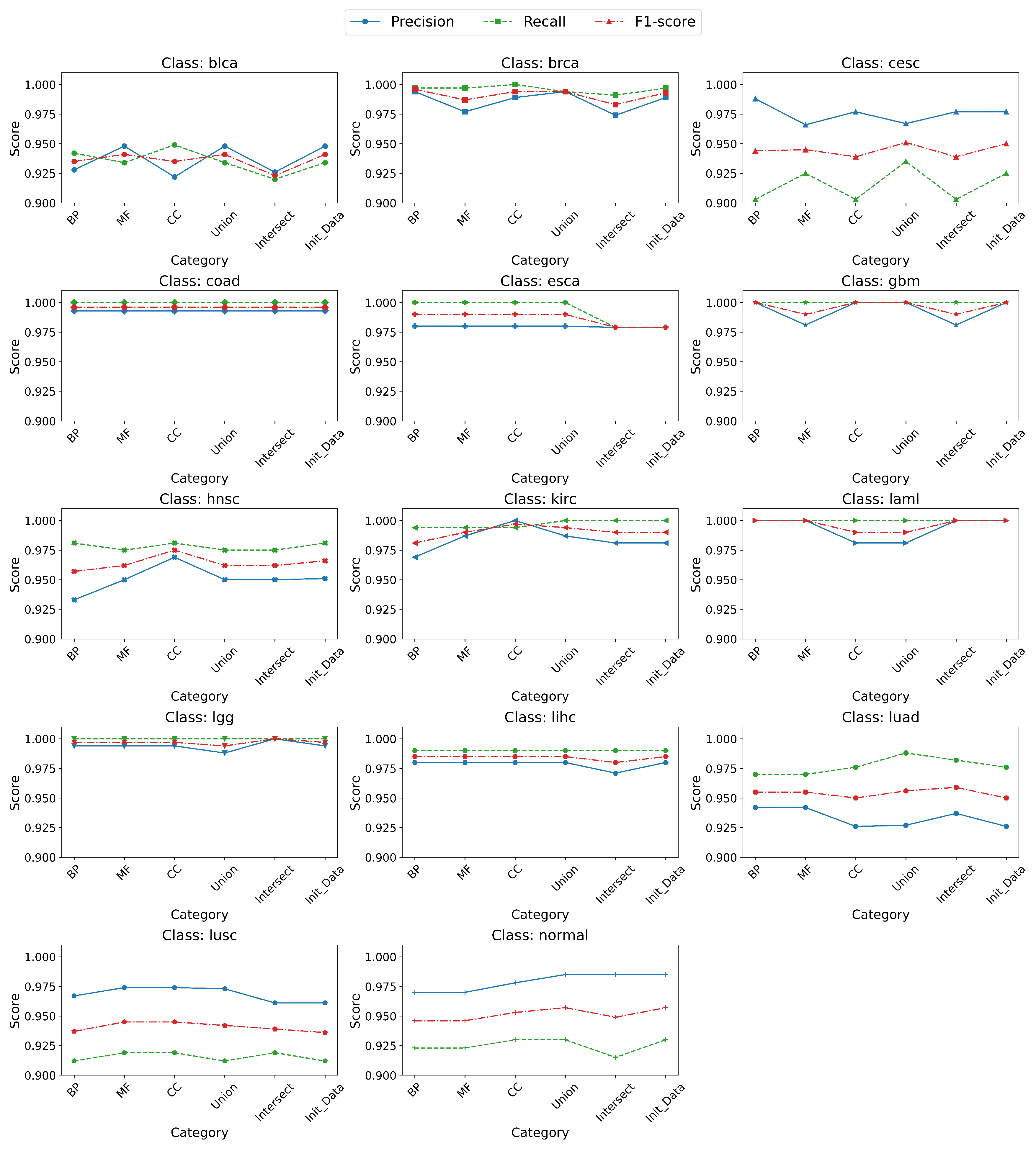
- Classification and Model Evaluation. To evaluate the discriminative potential of each constructed dataset, a supervised classification model is applied. A random forest classifier was selected due to its high performance on high-dimensional data, resistance to overfitting, and ability to estimate feature importance [25,26,27].Model training and evaluation are performed using a 70/30 stratified train–test split. Hyperparameter tuning was conducted exclusively on the training set using Bayesian optimization combined with 5-fold cross-validation to ensure generalizability and prevent information leakage. The independent test set was reserved for final performance evaluation using the optimized model.Bayesian optimization is a method that leverages probabilistic models (typically Gaussian processes) to iteratively select the most promising hyperparameter values based on a surrogate model of the objective function. Unlike random or grid search, Bayesian optimization strategically explores the hyperparameter space, reducing computational cost while improving convergence to optimal solutions. The objective function was defined as the average classification accuracy across validation folds during training.The following key hyperparameters are optimized as part of the modeling procedure: the number of decision trees in the ensemble (n_estimators); the maximum depth of individual trees (max_depth); the minimum number of samples required to split an internal node (min_samples_split); the minimum number of samples required to be at a leaf node (min_samples_leaf); the number of features considered when determining the best split (max_features); and the criterion for measuring the quality of a split, such as gini or entropy.Additionally, bootstrap sampling is enabled during model training to increase model variance and reduce the risk of overfitting.The trained model is evaluated on the test data using standard performance metrics, including precision, recall, F1-score, weighted F1-score, and accuracy, defined as follows:where , , , and denote true positives, false positives, true negatives, and false negatives, respectively; C is the number of classes; is the number of true samples in class i; and N is the total number of samples.These metrics were used to assess and compare the classification performance for each of the five gene subsets generated during the pipeline.
- 9.
- Analysis of Results. The classification results are analyzed to compare the predictive power of datasets based on individual GO categories and their combination. The impact of GO-based feature selection on classification performance is also evaluated.
3. Results and Discussion
3.1. Results of Data Preprocessing
3.2. Application of GO Analysis to Identify a Subset of Significant Genes
3.3. Quantitative Summary and Visualization of Gene Set Overlaps
3.4. Application of the Random Forest Classifier to the Generated Gene Expression Datasets
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GO | Gene Ontology |
| BP | Biological Process |
| MF | Molecular Function |
| CC | Cellular Component |
| T2DM | Type 2 Diabetes Mellitus |
| CRC | Colorectal Cancer |
| CHAF1B | Chromosomal Assembly Factor 1B |
| LUAD | Lung Adenocarcinoma |
| PPH | Postpartum Hemorrhage |
| DPYD | Dihydropyrimidine Dehydrogenase |
| DEGs | Differentially Expressed Genes |
| TCGA | The Cancer Genome Atlas |
| CPM | Counts Per Million |
| FDR | False Discovery Rate |
| BH | Benjamini–Hochberg |
| RF | Random Forest |
| SHAP | Shapley Additive Explanations |
References
- He, C.; Liu, M.; Hsiang, S.; Pierce, N.; Megahed, S.; Godfrey, A. An Ontological Knowledge-Driven Smart Contract Framework for Implicit Bridge Preservation Decision Making. J. Constr. Eng. Manag. 2025, 151, 04025008. [Google Scholar] [CrossRef]
- Geerts, G.; O’Leary, D. ORSO: The Organizational Structure Ontology. Account. Rev. 2025, 100, 261–290. [Google Scholar] [CrossRef]
- Saxena, R.; Bishnoi, R.; Singla, D. Gene Ontology: Application and importance in functional annotation of the genomic data. In Bioinformatics: Methods and Applications; Academic Press: Cambridge, MA, USA, 2021; pp. 145–157. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Combes, F.; Loux, V.; Vandenbrouck, Y. GO Enrichment Analysis for Differential Proteomics Using ProteoRE. Methods Mol. Biol. 2021, 2361, 179–196. [Google Scholar] [CrossRef] [PubMed]
- Hassan, H.; Shanak, S. GOTrapper: A tool to navigate through branches of gene ontology hierarchy. BMC Bioinform. 2019, 20, 20. [Google Scholar] [CrossRef]
- Paplińska-Goryca, M.; Misiukiewicz-Stępień, P.; Wróbel, M.; Mycroft-Rzeszotarska, K.; Adamska, D.; Rachowka, J.; Królikowska, M.; Goryca, K.; Krenke, R. The impaired response of nasal epithelial cells to microplastic stimulation in asthma and COPD. Sci. Rep. 2025, 15, 4242. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Zhu, Y.; Guo, Z.; Zheng, C.; Fu, X.; You, F.; Li, X. Exploring biomarkers and molecular mechanisms of Type 2 diabetes mellitus promotes colorectal cancer progression based on transcriptomics. Sci. Rep. 2025, 15, 4086. [Google Scholar] [CrossRef]
- Du, W.; Wu, X.W.; Li, Q.F.; Zhang, B.Y.; Wu, J.; Xu, Y.P.; Yi, X. Integrated bioinformatics and experimental analysis of CHAF1B as a novel biomarker and immunotherapy target in LUAD. Discov. Oncol. 2025, 16, 43. [Google Scholar] [CrossRef]
- Qu, J.; Jiang, H.; Shi, H.; Huang, N.; Su, J.; Zhang, Y.; Chen, L.; Zhao, Y. Novel predictive biomarkers for atonic postpartum hemorrhage as explored by proteomics and metabolomics. BMC Pregnancy Childbirth 2025, 25, 96. [Google Scholar] [CrossRef]
- Mahajan, P.; Fiehn, O.; Barupal, D. IDSL.GOA: Gene ontology analysis for interpreting metabolomic datasets. Sci. Rep. 2024, 14, 1299. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, Z.; Yang, Y.; Han, C.; Wang, L.; Kang, K.; Zhao, A. Exploration of potential novel drug targets and biomarkers for small cell lung cancer by plasma proteomescreening. Front. Pharmacol. 2023, 14, 1266782. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Yang, X.; Wu, W.; Yang, J. Identification of Novel Protein Biomarkers and Drug Targets for Acne Vulgaris by Integrating Human PlasmaProteome with Genome-Wide Association Data. J. Inflamm. Res. 2024, 17, 4431–4441. [Google Scholar] [CrossRef] [PubMed]
- Pomaznoy, M.; Ha, B.; Peters, B. GOnet: A tool for interactive Gene Ontology analysis. BMC Bioinform. 2018, 19, 470. [Google Scholar] [CrossRef]
- Hale, M.; Thapa, I.; Ghersi, D. FunSet: An open-source software and web server for performing and displaying Gene Ontology enrichmentanalysis. BMC Bioinform. 2019, 20, 359. [Google Scholar] [CrossRef]
- Volpato, M.; Hull, M.; Carr, I. GOTermViewer: Visualization of Gene Ontology Enrichment in Multiple Differential Gene Expression Analyses. Bioinform. Biol. Insights 2024, 18, 11779322241271550. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2024. [Google Scholar]
- Xu, S.; Hu, E.; Cai, Y.; Xie, Z.; Luo, X.; Zhan, L.; Tang, W.; Wang, Q.; Liu, B.; Wang, R.; et al. Using clusterProfiler to characterize multiomics data. Nat. Protoc. 2024, 19, 3292–3320. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Yan, G.R.; He, Q.Y. DOSE: An R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics 2015, 31, 608–609. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.I.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Carlson, M. org.Hs.eg.db: Genome Wide Annotation for Human. R Package Version 3.20.0. Bioconductor. 2024. Available online: https://bioconductor.org/packages/org.Hs.eg.db (accessed on 10 April 2025).
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. biomaRt: Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package. Bioinformatics 2009, 25, 526–528. [Google Scholar] [CrossRef]
- Haynes, W. Benjamini–Hochberg Method. In Encyclopedia of Systems Biology; Dubitzky, W., Wolkenhauer, O., Cho, K.H., Yokota, H., Eds.; Springer: New York, NY, USA, 2013; p. 78. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenführer, J.; Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 2006, 22, 1600–1607. [Google Scholar] [CrossRef]
- Wang, H.; Len, L.; Hu, L.; Hu, Y. Combining machine learning and single-cell sequencing to identify key immune genes in sepsis. Sci. Rep. 2025, 15, 1557. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.; Takaoka, E. An enhanced machine learning approach with stacking ensemble learner for accurate liver cancer diagnosis using feature selection and gene expression data. Healthc. Anal. 2025, 7, 100373. [Google Scholar] [CrossRef]
- Li, C.; Hao, R.; Li, C.; Liu, L.; Ding, Z. Integration of single-cell and bulk RNA sequencing data using machine learning identifies oxidative stress-related genes LUM and PCOLCE2 as potential biomarkers for heart failure. Int. J. Biol. Macromol. 2025, 300, 140793. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. Stats: The R Stats Package; R Package Version 4.3.1; R Core Team: Vienna, Austria, 2024. [Google Scholar]
- Gross, J.; Ligges, U. Nortest: Tests for Normality, R Package Version 1.0-4; 2022. Available online: https://CRAN.R-project.org/package=nortest (accessed on 10 April 2025).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Hyperparameter | Search Range | Description | Optimized Value |
|---|---|---|---|---|
| 1 | Number of Trees () | 10–200 | Total trees in the ensemble | 50 |
| 2 | Maximum Depth () | 1–20 | Limits tree depth to avoid overfitting | 14 |
| 3 | Min. Samples to Split () | 2–10 | Minimum number of samples to split a node | 9 |
| 4 | Min. Samples per Leaf () | 1–4 | Minimum number of samples in a terminal node | 4 |
| 5 | Max. Features () | 0.1–1.0 | Fraction of features used for splitting | 0.113 |
| 6 | Bootstrap Sampling () | {True, False} | Enables sampling with replacement | True |
| 7 | Split Criterion () | {Gini, entropy} | Function to measure split quality | Entropy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babichev, S.; Yarema, O.; Liakh, I.; Shumylo, N. A Gene Ontology-Based Pipeline for Selecting Significant Gene Subsets in Biomedical Applications. Appl. Sci. 2025, 15, 4471. https://doi.org/10.3390/app15084471
Babichev S, Yarema O, Liakh I, Shumylo N. A Gene Ontology-Based Pipeline for Selecting Significant Gene Subsets in Biomedical Applications. Applied Sciences. 2025; 15(8):4471. https://doi.org/10.3390/app15084471
Chicago/Turabian StyleBabichev, Sergii, Oleg Yarema, Igor Liakh, and Nataliia Shumylo. 2025. "A Gene Ontology-Based Pipeline for Selecting Significant Gene Subsets in Biomedical Applications" Applied Sciences 15, no. 8: 4471. https://doi.org/10.3390/app15084471
APA StyleBabichev, S., Yarema, O., Liakh, I., & Shumylo, N. (2025). A Gene Ontology-Based Pipeline for Selecting Significant Gene Subsets in Biomedical Applications. Applied Sciences, 15(8), 4471. https://doi.org/10.3390/app15084471