Silicon-[18F]Fluorine Radiochemistry: Basics, Applications and Challenges


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
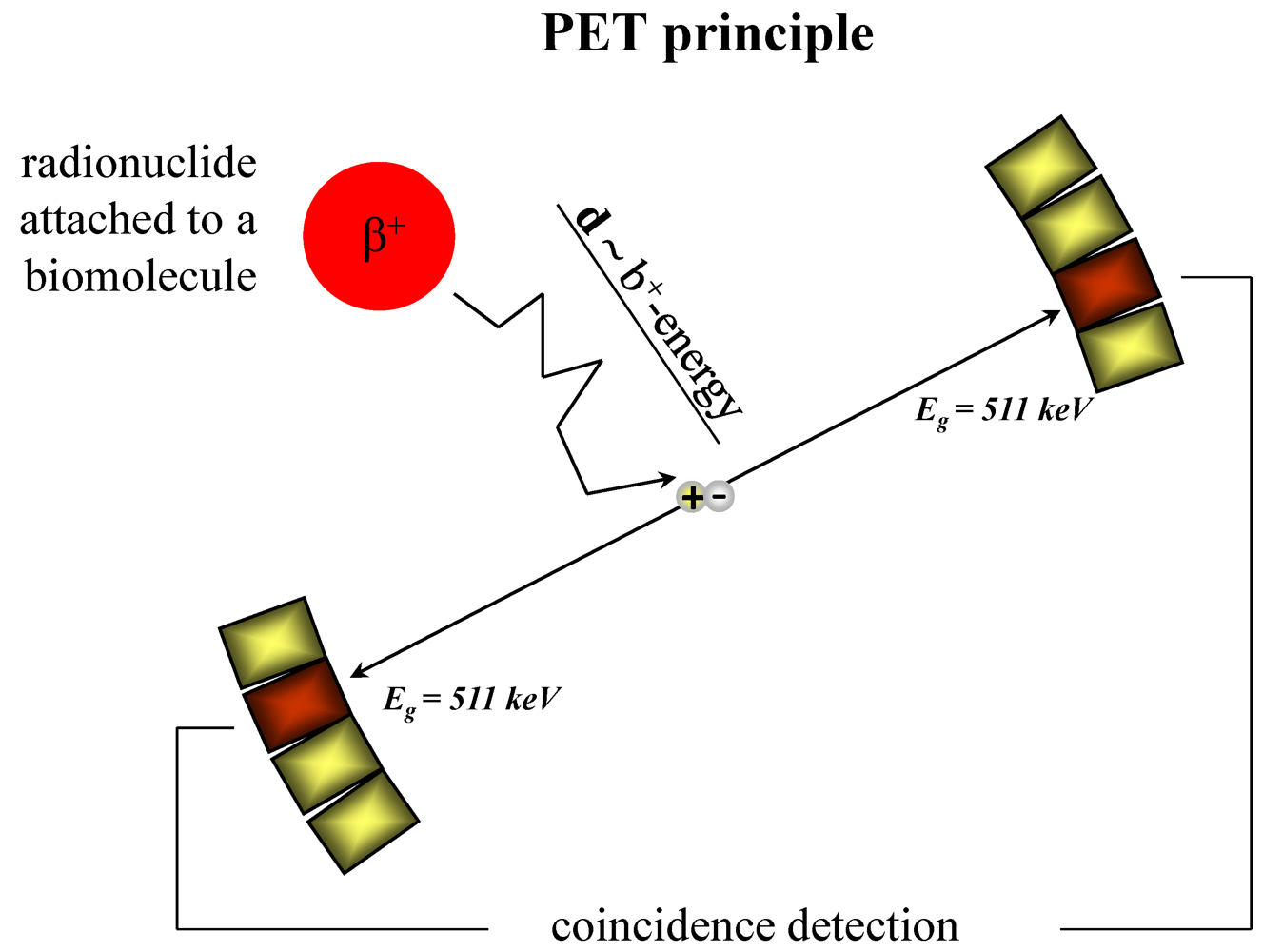
:1. Introduction: Fluorine-18, an Important Nuclide for PET Imaging

2. Basics of Silicon-18F Radiochemistry
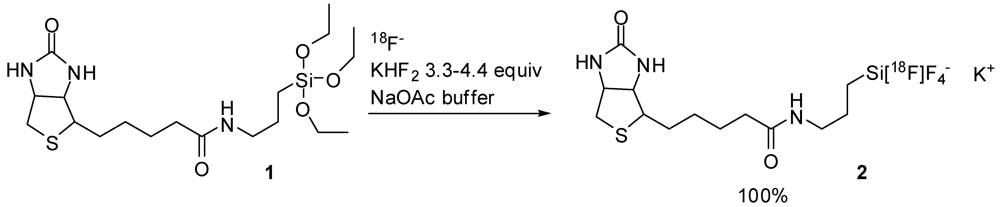
3. [18F]Tetrafluorosiliconates “Ate” Salts

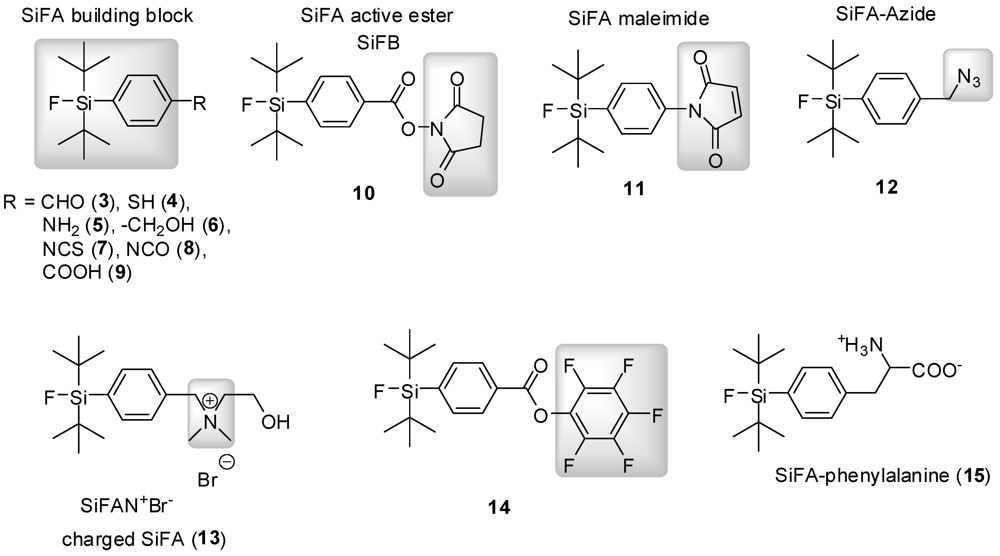
4. Silicon-Fluoride-Acceptors (SiFAs): An introduction

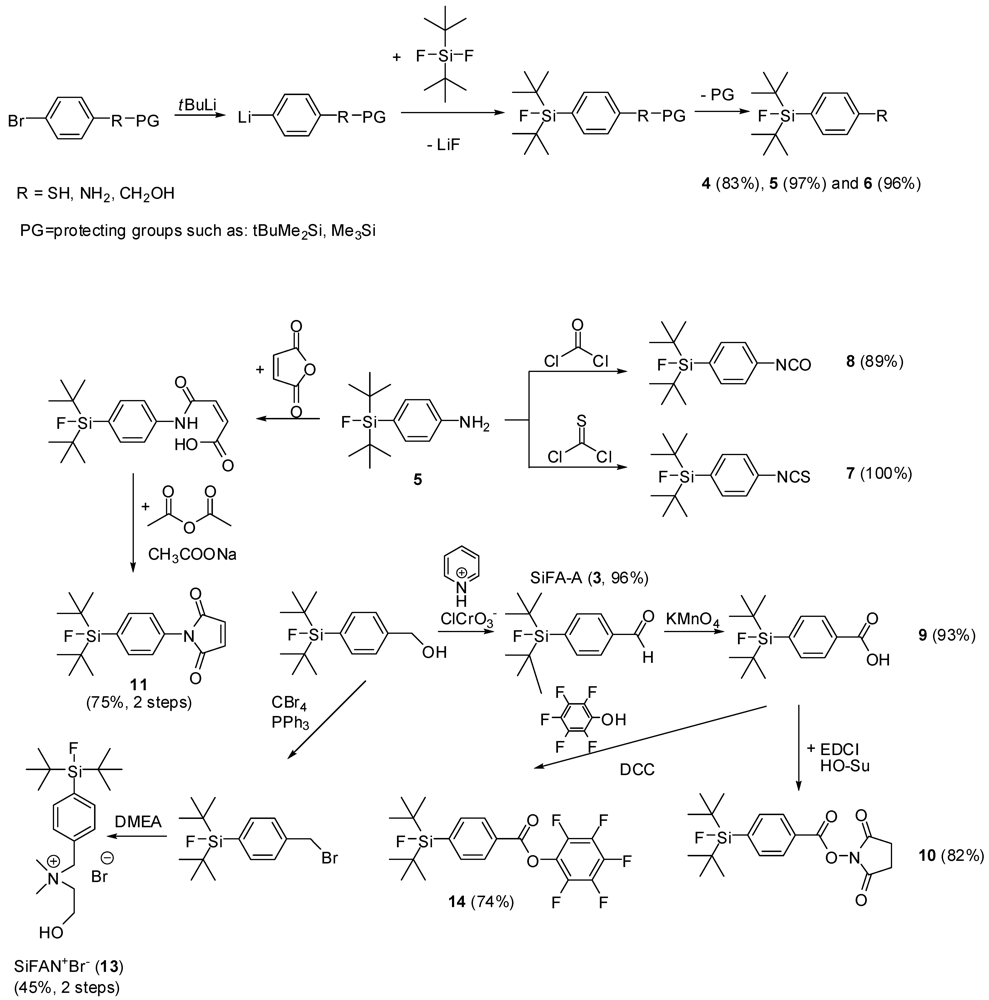
5. The synthesis of SiFAs

6. SiFAs and Their High Lipophilicity

7. Silicon Building Blocks Containing H, OH and Alkoxy Moieties as Leaving Groups



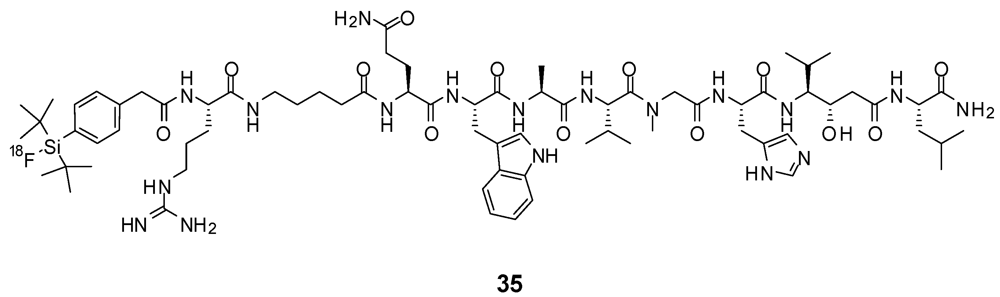
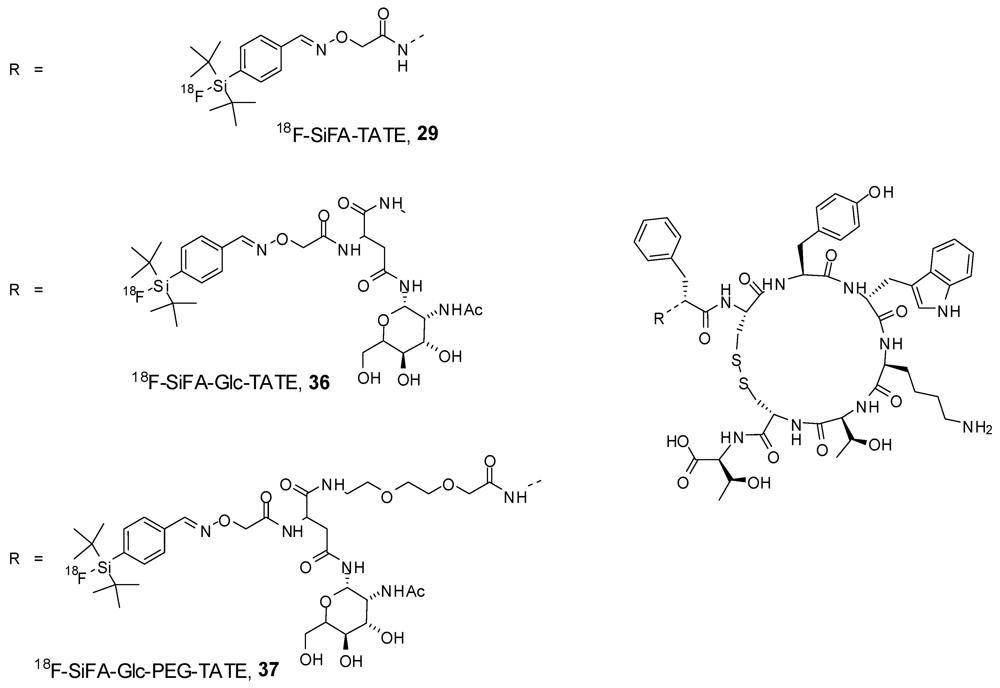
8. Peptides




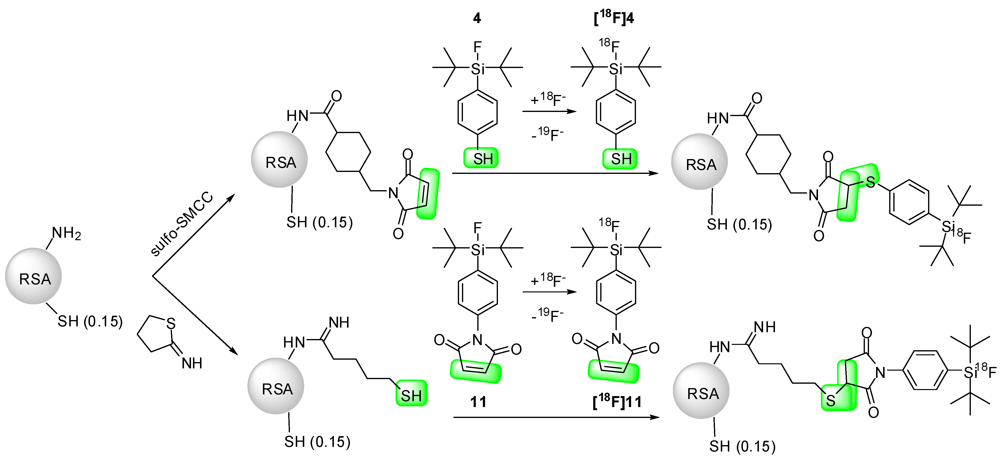
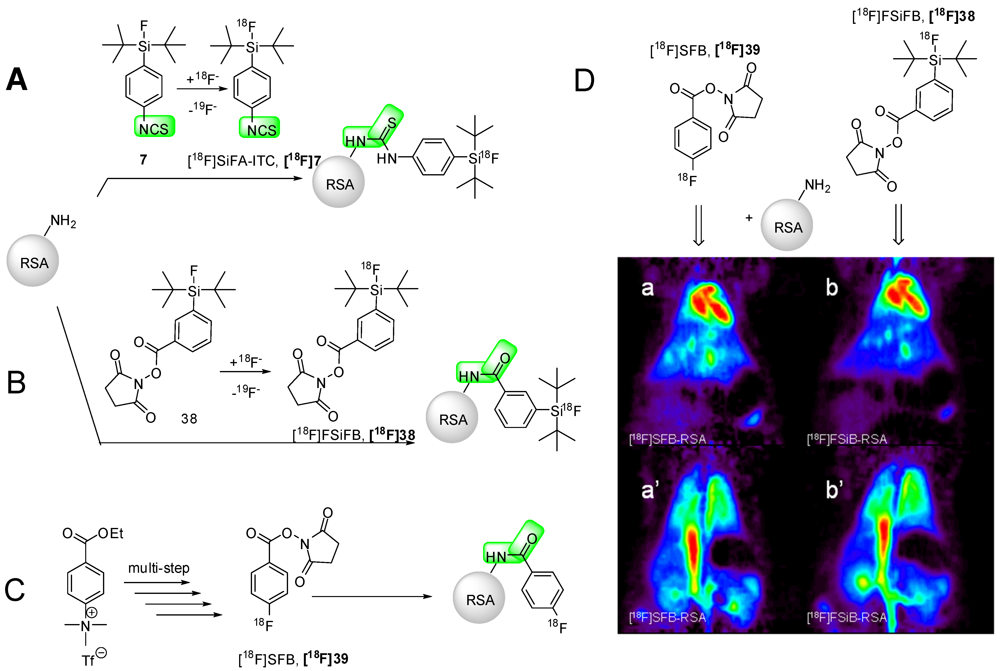
9. Protein Labeling


10. Small Molecules



11. Conclusions
Conflict of Interest
References
- Wahl, R.L.; Buchanan, J.W. Principles and Practice of Positron Emission Tomography; Lippincott Williams & Wilkins: Philadelphia, USA, 2002. [Google Scholar]
- Cai, L.S.; Lu, S.Y.; Pike, V.W. Chemistry with [18F]fluoride ion. Eur. J. Org. Chem. 2008, 2853–2873. [Google Scholar]
- Schubiger, P.A.; Lehmann, L.; Friebe, M. Chemistry—The Driving Force in Molecular Imaging; Springer: Berlin Heidelberg, Germay, 2007; Volume 97, pp. 1291–1297. [Google Scholar]
- Wester, H.J. Munich Molecular Imaging Series; Scintomics: Fürstenfeldbruck, Germany, 2010; Volume 1, pp. 5–74. [Google Scholar]
- Ting, R.; Adam, M.J.; Ruth, T.J.; Perrin, D.M. Arylfluoroborates and alkylfluorosilicates as potential PET imaging agents: High-yielding aqueous biomolecular F-18-labeling. J. Am. Chem. Soc. 2005, 127, 13094–13095. [Google Scholar]
- Ting, R.; Harwig, C.; auf dem Keller, U.; McCormick, S.; Austin, P.; Overall, C.M.; Adam, M.J.; Ruth, T.J.; Perrin, D.M. Toward [18F]-labeled aryltrifluoroborate radiotracers: In vivo Positron Emission Tomography imaging of stable aryltrifluoroborate clearance in mice. J. Am. Chem. Soc. 2008, 130, 12045–12055. [Google Scholar]
- Keller, U.A.D.; Bellac, C.L.; Li, Y.; Lou, Y.M.; Lange, P.F.; Ting, R.; Harwig, C.; Kappelhoff, R.; Dedhar, S.; Adam, M.J.; Ruth, T.J.; Benard, F.; Perrin, D.M.; Overall, C.M. Novel matrix metalloproteinase inhibitor [18F]marimastat-aryltrifluoroborate as a probe for in vivo Positron Emission Tomography imaging in cancer. Cancer Res. 2010, 70, 7562–7569. [Google Scholar]
- McBride, W.J.; D’Souza, C.A.; Sharkey, R.M.; Karacay, H.; Rossi, E.A.; Chang, C.H.; Goldenberg, D.M. Improved 18F labeling of peptides with a fluoride-aluminum-chelate complex. Bioconjugate Chem. 2010, 21, 1331–1340. [Google Scholar] [CrossRef]
- McBride, W.J.; Sharkey, R.M.; Karacay, H.; D'Souza, C.A.; Rossi, E.A.; Laverman, P.; Chang, C.H.; Boerman, O.C.; Goldenberg, D.M. A novel method of 18F radiolabeling for PET. J. Nucl. Med. 2009, 50, 991–998. [Google Scholar] [CrossRef]
- Laverman, P.; McBride, W.J.; Sharkey, R.M.; Eek, A.; Joosten, L.; Oyen, W.J.G.; Goldenberg, D.M.; Boerman, O.C. A novel facile method of labeling octreotide with 18F-fluorine. J. Nucl. Med. 2010, 51, 454–461. [Google Scholar] [CrossRef]
- McBride, W.J.; D’Souza, C.A.; Karacay, H.; Sharkey, R.M.; Goldenberg, D.M. New lyophilized kit for rapid radiofluorination of peptides. Bioconjugate Chem. 2012, 23, 538–547. [Google Scholar] [CrossRef]
- McBride, W.J.; D’Souza, C.A.; Sharkey, R.M.; Goldenberg, D.M. The radiolabeling of proteins by the [18F]AlF method. Appl. Radiat. Isotopes 2012, 70, 200–204. [Google Scholar] [CrossRef]
- Mu, L.; Schubiger, A.P.; Ametamey, S.M. [18F]Fluorosilicon- and [18F]Fluoroboron-based biomolecules for PET imaging. Curr. Radiophar. 2010, 3, 224–242. [Google Scholar] [CrossRef]
- Ametamey, S.M.; Honer, M.; Schubiger, P.A. Molecular imaging with PET. Chem. Rev. 2008, 108, 1501–1516. [Google Scholar] [CrossRef]
- Schirrmacher, R.; Wangler, C.; Schirrmacher, E. Recent developments and trends in 18F-radiochemistry: Syntheses and applications. Mini-Rev. Org. Chem. 2007, 4, 317–329. [Google Scholar] [CrossRef]
- Smith, G.E.; Sladen, H.L.; Biagini, S.C.G.; Blower, P.J. Inorganic approaches for radiolabelling biomolecules with fluorine-18 for imaging with Positron Emission Tomography. Dalton Trans 2011, 40, 6196–6205. [Google Scholar] [CrossRef]
- Miller, P.W.; Long, N.J.; Vilar, R.; Gee, A.D. Synthesis of C-11, F-18, O-15, and N-13 radiolabels for Positron Emission Tomography. Angew Chem. Int. Ed. 2008, 47, 8998–9033. [Google Scholar] [CrossRef]
- Mamat, C.; Ramenda, T.; Wuest, F.R. Recent applications of click chemistry for the synthesis of radiotracers for molecular imaging. Mini-Rev. Org. Chem. 2009, 6, 21–34. [Google Scholar] [CrossRef]
- Gens, T.A.; Wethington, J.A.; Brosi, A.R. The exchange of F-18 between metallic fluorides and silicon tetrafluoride. J. Phys. Chem.-Us 1959, 62, 1593–1593. [Google Scholar]
- Poole, R.T.; Winfield, J.M. Radiotracers in fluorine chemistry. 4. F-18 exchange between labeled alkylfluorosilanes and fluorides, or fluoride methoxides, of Tungsten(Vi), Molybdenum(Vi), Tellurium(Vi), and Iodine(V). J. Chem. Soc. Dalton 1976, 1557–1560. [Google Scholar]
- Winfield, J.M. Preparation and use of 18-fluorine labeled inorganic-compounds. J. Fluorine Chem. 1980, 16, 1–17. [Google Scholar] [CrossRef]
- Sanyal, D.K.; Winfield, J.M. Radiotracers in fluorine chemistry. 8. F-18 exchange-reactions involving Uranium(VI) or Uranium(V) fluorides—Evidence for surface complexation and comparisons with ligand-exchange reactions. J. Fluorine Chem. 1984, 24, 75–92. [Google Scholar] [CrossRef]
- Rosenthal, M.S.; Bosch, A.L.; Nickles, R.J.; Gatley, S.J. Synthesis and some characteristics of no-carrier added [18F]fluorotrimethylsilane. Int. J. Appl. Radiat. Isotop. 1985, 36, 318–319. [Google Scholar] [CrossRef]
- Gatley, S.J. Rapid production and trapping of [18F] Fluorotrimethylsilane, and its use in nucleophilic fluorine-18 labeling without an aqueous evaporation step. Appl. Radiat. Isotope. 1989, 40, 541–544. [Google Scholar] [CrossRef]
- Hamacher, K.; Coenen, H.H.; Stocklin, G. Efficient stereospecific synthesis of no-carrier-added 2-[18F]-Fluoro-2-deoxy-D-glucose using aminopolyether supported nucleophilic-substitution. J. Nucl. Med. 1986, 27, 235–238. [Google Scholar]
- Walsh, J.C.; Fleming, L.M.; Satyamurthy, N.; Barrio, J.R.; Phelps, M.E.; Gambhir, S.S.; Toyokuni, T. Application of silicon-fluoride chemistry for the development of amine-reactive F-18-labeling agents for biomolecules. J. Nucl. Med 2000, 41, 249p–249p. [Google Scholar]
- Choudhry, U.; Martin, K.E.; Biagini, S.; Blower, P.J. Alkoxysilane groups for instant labeling of biomolecules with 18F. Nucl. Med. Commun. 2006, 27. [Google Scholar]
- Schirrmacher, R.; Bradtmöller, G.; Schirrmacher, E.; Thews, O.; Tillmanns, J.; Siessmeier, T.; Buchholz, H.G.; Bartenstein, P.; Wängler, B.; Niemeyer, C.M.; Jurkschat, K. 18F-labeling of peptides by means of an organosilicon-based fluoride acceptor. Angew Chem. Int. Ed. 2006, 45, 6047–6050. [Google Scholar]
- Mu, L.J.; Hohne, A.; Schubiger, R.A.; Ametamey, S.M.; Graham, K.; Cyr, J.E.; Dinkelborg, L.; Stellfeld, T.; Srinivasan, A.; Voigtmann, U.; Klar, U. Silicon-based building blocks for one-step 18F-radiolabeling of peptides for PET imaging. Angew Chem. Int. Ed. 2008, 47, 4922–4925. [Google Scholar]
- Schirrmacher, E.; Wangler, B.; Cypryk, M.; Bradtmoller, G.; Schafer, M.; Eisenhut, M.; Jurkschat, K.; Schirrmacher, R. Synthesis of p-(Di-tert-butyl[18F]fluorosilyl)benzaldehyde ([18F]SiFA-A) with high specific activity by isotopic exchange: A convenient Labeling synthon for the 18F-labeling of n-amino-oxy derivatized peptides. Bioconjugate Chem. 2007, 18, 2085–2089. [Google Scholar] [CrossRef]
- Tietze, L.F.; Schmuck, K. SiFA azide: A new building block for PET imaging using click chemistry. Synlett 2011, 1697–1700. [Google Scholar] [CrossRef]
- Kostikov, A.P.; Iovkova, L.; Chin, J.; Schirrmacher, E.; Wangler, B.; Wangler, C.; Jurkschat, K.; Cosa, G.; Schirrmacher, R. N-(4-(di-tert-butyl[(18)F]fluorosilyl)benzyl)-2-hydroxy-N, N-dimethylethylammonium bromide ([18F]SiFAN+Br−): A novel lead compound for the development of hydrophilic SiFA-based prosthetic groups for (18)F-labeling. J. Fluorine Chem. 2011, 132, 27–34. [Google Scholar] [CrossRef]
- Hohne, A.; Yu, L.; Mu, L.J.; Reiher, M.; Voigtmann, U.; Klar, U.; Graham, K.; Schubiger, P.A.; Ametamey, S.M. Organofluorosilanes as model compounds for 18F-labeled Silicon-based PET tracers and their hydrolytic stability: Experimental data and theoretical calculations (PET = Positron Emission Tomography). Chem. Eur. J. 2009, 15, 3736–3743. [Google Scholar]
- Kiesewetter, D.O.; Jacobson, O.; Lang, L.X.; Chen, X.Y. Automated radiochemical synthesis of [18F]FBEM: A thiol reactive synthon for radiofluorination of peptides and proteins. Appl. Radiat. Isotopes 2011, 69, 410–414. [Google Scholar] [CrossRef]
- Speranza, A.; Ortosecco, G.; Castaldi, E.; Nardelli, A.; Pace, L.; Salvatore, M. Fully automated synthesis procedure of 4-[18F]fluorobenzaldehyde by commercial synthesizer: Amino-oxi peptide labelling prosthetic group. Appl. Radiat. Isotopes 2009, 67, 1664–1669. [Google Scholar] [CrossRef]
- Thonon, D.; Goblet, D.; Goukens, E.; Kaisin, G.; Paris, J.; Aerts, J.; Lignon, S.; Franci, X.; Hustinx, R.; Luxen, A. Fully automated preparation and conjugation of N-succinimidyl 4-[18F]fluorobenzoate ([18F]SFB) with RGD peptide using a GE FASTlab (TM) synthesizer. Mol. Imaging Biol. 2011, 13, 1088–1095. [Google Scholar] [CrossRef]
- D'Souza, C.A.; McBride, W.J.; Sharkey, R.M.; Todaro, L.J.; Goldenberg, D.M. High-yielding aqueous 18F-labeling of peptides via Al18F chelation. Bioconjugate Chem. 2011, 22, 1793–1803. [Google Scholar]
- Wängler, C.; Waser, B.; Alke, A.; Iovkova, L.; Buchholz, H.G.; Niedermoser, S.; Jurkschat, K.; Fottner, C.; Bartenstein, P.; Schirrmacher, R.; Reubi, J.C.; Wester, H.J.; Wängler, B. One-step 18F-labeling of carbohydrate-conjugated octreotate-derivatives containing a Silicon-Fluoride-Acceptor (SiFA): In vitro and in vivo evaluation as tumor imaging agents for Positron Emission Tomography (PET). Bioconjugate Chem. 2010, 21, 2289–2296. [Google Scholar]
- Iovkova, L.; Konning, D.; Wangler, B.; Schirrmacher, R.; Schoof, S.; Arndt, H.D.; Jurkschat, K. SiFA-modified phenylalanine: A key compound for the efficient synthesis of 18F-labelled peptides. Eur. J. Inorg. Chem. 2011, 2238–2246. [Google Scholar]
- Höhne, A.; Mu, L.; Honer, M.; Schubiger, P.A.; Ametamey, S.M.; Graham, K.; Stellfeld, T.; Borkowski, S.; Berndorff, D.; Klar, U.; Voigtmann, U.; Cyr, J.E.; Friebe, M.; Dinkelborg, L.; Srinivasan, A. Synthesis, 18F-labeling, and in vitro and in vivo studies of bombesin peptides modified with silicon-based building blocks. Bioconjugate Chem. 2008, 19, 1871–1879. [Google Scholar] [CrossRef]
- Balentova, E.; Collet, C.; Lamande-Langle, S.; Chretien, F.; Thonon, D.; Aerts, J.; Lemaire, C.; Luxen, A.; Chapleur, Y. Synthesis and hydrolytic stability of novel 3-[[18F]fluoroethoxybis (1-methylethyl)silyl]propanamine-based prosthetic groups. J. Fluorine Chem. 2011, 132, 250–257. [Google Scholar] [CrossRef]
- Schottelius, M.; Rau, F.; Reubi, J.C.; Schwaiger, M.; Wester, H.A. Modulation of pharmacokinetics of radioiodinated sugar-conjugated somatostatin analogues by variation of peptide net charge and carbohydration chemistry. Bioconjugate Chem. 2005, 16, 429–437. [Google Scholar] [CrossRef]
- Schottelius, M.; Wester, H.J.; Reubi, J.C.; Senekowitsch-Schmidtke, R.; Schwaiger, M. Improvement of pharmacokinetics of radioiodinated Tyr3-octreotide by conjugation with carbohydrates. Bioconjugate Chem. 2002, 13, 1021–1030. [Google Scholar] [CrossRef]
- Antunes, P.; Ginj, M.; Walter, M.A.; Chen, J.H.; Reubi, J.C.; Maecke, H.R. Influence of different spacers on the biological profile of a DOTA-somatostatin analogue. Bioconjugate Chem. 2007, 18, 84–92. [Google Scholar] [CrossRef]
- Rosa-Neto, P.; Wängler, B.; Iovkova, L.; Boening, G.; Reader, A.; Jurkschat, K.; Schirrmacher, E. [18F]SiFA-isothiocyanate: A new highly effective radioactive labeling agent for lysine-containing proteins. Chembiochem 2009, 10, 1321–1324. [Google Scholar] [CrossRef]
- Bohn, P.; Deyine, A.; Azzouz, R.; Bailly, L.; Fiol-Petit, C.; Bischoff, L.; Fruit, C.; Marsais, F.; Vera, P. Design of silicon-based misonidazole analogues and 18F-radiolabelling. Nucl. Med. Biol. 2009, 36, 895–905. [Google Scholar] [CrossRef]
- Schulz, J.; Vimont, D.; Bordenave, T.; James, D.; Escudier, J.M.; Allard, M.; Szlosek-Pinaud, M.; Fouquet, E. Silicon-based chemistry: An original and efficient one-step approach to [18F]-nucleosides and [18F]-oligonucleotides for PET imaging. Chem. Eur. J. 2011, 17, 3096–3100. [Google Scholar]
- Iovkova-Berends, L.; Wangler, C.; Zoller, T.; Hofner, G.; Wanner, K.T.; Rensch, C.; Bartenstein, P.; Kostikov, A.; Schirrmacher, R.; Jurkschat, K.; Wangler, B. t-Bu2SiF-derivatized D2-receptor ligands: The first SiFA-containing small molecule radiotracers for target-specific PET-imaging. Molecules 2011, 16, 7458–7479. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wängler, C.; Kostikov, A.; Zhu, J.; Chin, J.; Wängler, B.; Schirrmacher, R. Silicon-[18F]Fluorine Radiochemistry: Basics, Applications and Challenges. Appl. Sci. 2012, 2, 277-302. https://doi.org/10.3390/app2020277
Wängler C, Kostikov A, Zhu J, Chin J, Wängler B, Schirrmacher R. Silicon-[18F]Fluorine Radiochemistry: Basics, Applications and Challenges. Applied Sciences. 2012; 2(2):277-302. https://doi.org/10.3390/app2020277
Chicago/Turabian StyleWängler, Carmen, Alexey Kostikov, Jun Zhu, Joshua Chin, Björn Wängler, and Ralf Schirrmacher. 2012. "Silicon-[18F]Fluorine Radiochemistry: Basics, Applications and Challenges" Applied Sciences 2, no. 2: 277-302. https://doi.org/10.3390/app2020277
APA StyleWängler, C., Kostikov, A., Zhu, J., Chin, J., Wängler, B., & Schirrmacher, R. (2012). Silicon-[18F]Fluorine Radiochemistry: Basics, Applications and Challenges. Applied Sciences, 2(2), 277-302. https://doi.org/10.3390/app2020277