1. Introduction
Zirconium-89 (
89Zr,
t1/2 = 78.4 h) decays via both β
+ emission (22.7%) and electron capture (73.3%) to an intermediate
89mY state (
t1/2 = 15.7 s), which decays to stable
89Y via a γ-ray emission of 909 keV. The long half-life of this radionuclide is well suited for the design of radiotracers, such as nanoparticules and monoclonal antibodies (mAb), which require extended in vivo circulation times for optimal biodistribution and tumour targeting [
1,
2,
3].
89Zr-based imaging presents numerous advantages, including a sufficiently high abundance of β
+ emission with low maximum energy (
Emax (β
+) = 897 keV and
Eave. (β
+) = 396.9 keV), resulting in a good spatial resolution for mAb-tracking by PET imaging [
4,
5,
6]. The most widespread application for
89Zr has been the development of immuno-PET tracers based on mAb for in vivo PET cancer imaging.
89Zr-immuno PET can significantly improve staging, provide an effective method to detect recurrence, and allow for the identification of patients who will be eligible for more personalized and adapted treatment to their specific cancer in order to improve their quality of life.
Zirconium-89 can be produced by cyclotron using three different approaches involving either proton (H
+), deuteron (
2H), or alpha (α) particle bombardment [
7,
8,
9,
10,
11]. The production yield of
89Zr obtained by (p,n) reaction on
89Y is comparable to the one of (p,pxn) reaction on
natZr and notably higher than (α,n) reaction on
natSr and (d,2n) reaction on
89Y, along with higher radionuclide purity [
9,
11,
12].
89Y is available under various forms, including foils, wires, and powder with purity greater than 99.6%. The target material is mononuclidic and the use of enriched isotopes for irradiation is unnecessary.
89Y foil is commonly chosen for its availability and easy handling [
13,
14]. High effective molar activity (EMA) of the isolated
89Zr fractions has been measured while using
89Y-foil [
5]. Zirconium-89 can also be produced using a liquid target with yttrium nitrate salts via the
89Y(p,n)
89Zr reaction. Production of radiometals in solution is challenging due to in-target salt precipitation and unstable target pressures that are caused by gas evolution during irradiation [
15]. Moreover, the production yield of
89Zr in solution target is still well below the solid target production yield [
5,
16]. Zweit et al. showed that the production of no-carrier-added
89Zr from deuteron irradiation of natural
89Y-pressed targets was obtained in high yield, but within the energy window studied,
88Zr was also produced [
8]. Other methods, including yttrium-sputtering deposition [
17] and spot-welding [
18], have been studied for the preparation of
89Zr, but pressing would be advantageous in view of cost and productivity.
In the current study, we investigated for the first time the use of an 89Y pressed powder target to produce 89Zr via proton irradiation. A comparative study with existing 89Y-foil target is presented in order to outline the full potential of this novel pressed target. We studied the dimension and thickness of the solid targets under different irradiation conditions and we optimized the design of the target supports. We also developed an automated extraction and purification process while using a cassette-based module allowing for the fractionation of 89Zr. Results are presented and discussed with the objective of maximizing the thickness of the solid targets and obtaining high-yield, high-purity, and high-EMA as 89Zr-oxalate.
2. Materials and Methods
2.1. Materials
All the chemicals and solvents were purchased with high purity and were used as is unless otherwise specified. Yttrium-89 foil (0.127 and 0.25-mm thick, 99.9%) and yttrium-89 powder 40–mesh (99.6%) with different metal composition (
Table 1) were bought from Alfa Aesar (Ward Hill, MA, USA/VWR International, CA).
Hydrochloric acid (99.999%), oxalic acid (99.999%), Na2CO3 (99.999%) trace metal basis, sodium hydroxide pellets (≥98.0%), diethylenetriaminepentaacetic acid (DTPA, ≥99%), and deferoxamine (DFO) mesylate salt (≥92.5%) were purchased from Sigma-Aldrich (Saint-Louis, MO, USA). High-purity water (Optima LC/MS, ultra-high performance liquid chromatography ultraviolet grade, 0.03-mm filtered) and acetonitrile (HPLC grade, 99.9%) were purchased from Fisher Scientific (Ottawa, ON, CA). All the glass vials were cleaned while using chromic sulfuric acid (Fisher Scientific, Ottawa, ON, CA). Instant thin layer chromatography-silica gel (ITLC-SG) was acquired from Agilent Technology (Santa Clara, CA, USA). The labeling efficiency of 89Zr-DFO was determined while using radio-ITLC-SG with 100 mM DTPA as the mobile phase. The radio-ITLC plates were scanned using an instant imager scanner (Bioscan, DC, USA). Radioactivity measurements were performed in an ionization chamber (CRC-25PET; Capintec) on the 89Zr setting to control process efficiency and by γ-ray spectrometry with a calibrated high-purity germanium detector (GMX HPGe; ORTEC, Oak Ridge, TN, USA) for analytic quantitation. Experimental samples were counted for 10 min by using dynamic energy windows of 1-2000 keV. All of the radioactive detection devices were calibrated and maintained in accordance with our routine quality control procedures.
2.2. Cyclotron Targetry and Irradiation
Irradiations were performed using TR19 and TR24 variable energy (H
–: 13–19 and 16.2–24 MeV, respectively) cyclotrons (ACSI, Richmond, BC, Canada) while using a straight 90° target station (ACSI).
89Zr was produced via the
89Y(p,n)
89Zr transmutation reaction while using a solid
89Y-foil or pressed target mounted on custom-made aluminum and niobium coin target supports (
Figure 1). Irradiation times (T
irr) were 1–5 h with a beam current of 10–40 μA for both cyclotrons. The beam size in all irradiation experiments was ~10 mm in diameter. The incident proton-beam energies were selected based on the degrader materials used and varied between 16.2–18 MeV. The proton-beam energies deposited on the target materials were 11.3–15 MeV for all the productions and they were calculated by Monte Carlo simulation while using SRIM [
19]. The ACSI small solid target irradiation station provided efficient target cooling through a helium gas jet directed on the front side of the target while the backside was cooled by chilled water (16.5–18.5 °C).
2.3. Solid Target Design and Preparation
Four aluminum and niobium coin target holders/degraders were custom-designed and manufactured at the Department of Physics’s machine shop of the Université de Sherbrooke (
Figure 1). Both aluminum and niobium coins have an outer diameter of 24 mm and a thickness of 2 mm to fit in the small solid target station, which can accommodate no more than 2.2-mm thick targets. Initially, the first target body Al-21-1, presenting a hole diameter of 21 mm and a thickness of 1 mm, was made from 6061-T6 aluminum alloy to accommodate a thickness of 0.254 mm of
89Y-foil at maximum (
Figure 1A). As the proton beam size of both cyclotrons is ~10 mm, the hole diameter of the niobium degraders was reduced to 13 mm (Nb-13-0.35) and 6 mm (Nb-6-0.35) to accommodate a 1-mm thick
89Y-foil/pressed powder (
Figure 1B,C). An aluminum open window holder with a hole diameter of 13 mm (Al-13) was also designed to be used with a degrader (
Figure 1D). As shown in
Figure 1B–D, small aluminum disks (13 and 6 mm diameter) were used when needed to fill the gap between
89Y-pressed or foil targets and the aluminum back support for improvement of targets cooling during the irradiation. The aluminum back supports were machined with a thickness of 0.5 mm for Al-13 holders when compared to a thickness of 1.0 mm for all other degraders. Target assembly was performed without applying pressure on target material, foil, or pellet. The procedure to assemble the targets is illustrated in
Figure S1 of the Supplementary Information.
Before pressing, inert gas (N
2) was applied into the hole in the die. The
89Y powder was inserted and pressed into either 6 or 13 mm diameter pellets ranging in thickness from 0.3 to 0.5 mm. The maximum pressure applied to 6 and 13 mm dies was 3500 and 18,000 pounds per square inch, respectively, while using a digital hydraulic carver press (Module number: 3912, Carver, Inc, Wabash, IN, US). The pellets were placed in the target holders and covered with a degrader.
89Y-foil targets of various thicknesses ranging from 0.25 to 0.51 mm were mounted on the custom-made holders (
Figure S1). Niobium and aluminum degraders were installed on the He/beam side to avoid problems, while mounting and dismounting the disc set in the cyclotron solid target station. For the optimization of the production yield, seven 0.89-mm thick
89Y foils were mounted together in a round shaped 6-mm diameter Nb-6-0.35 degrader.
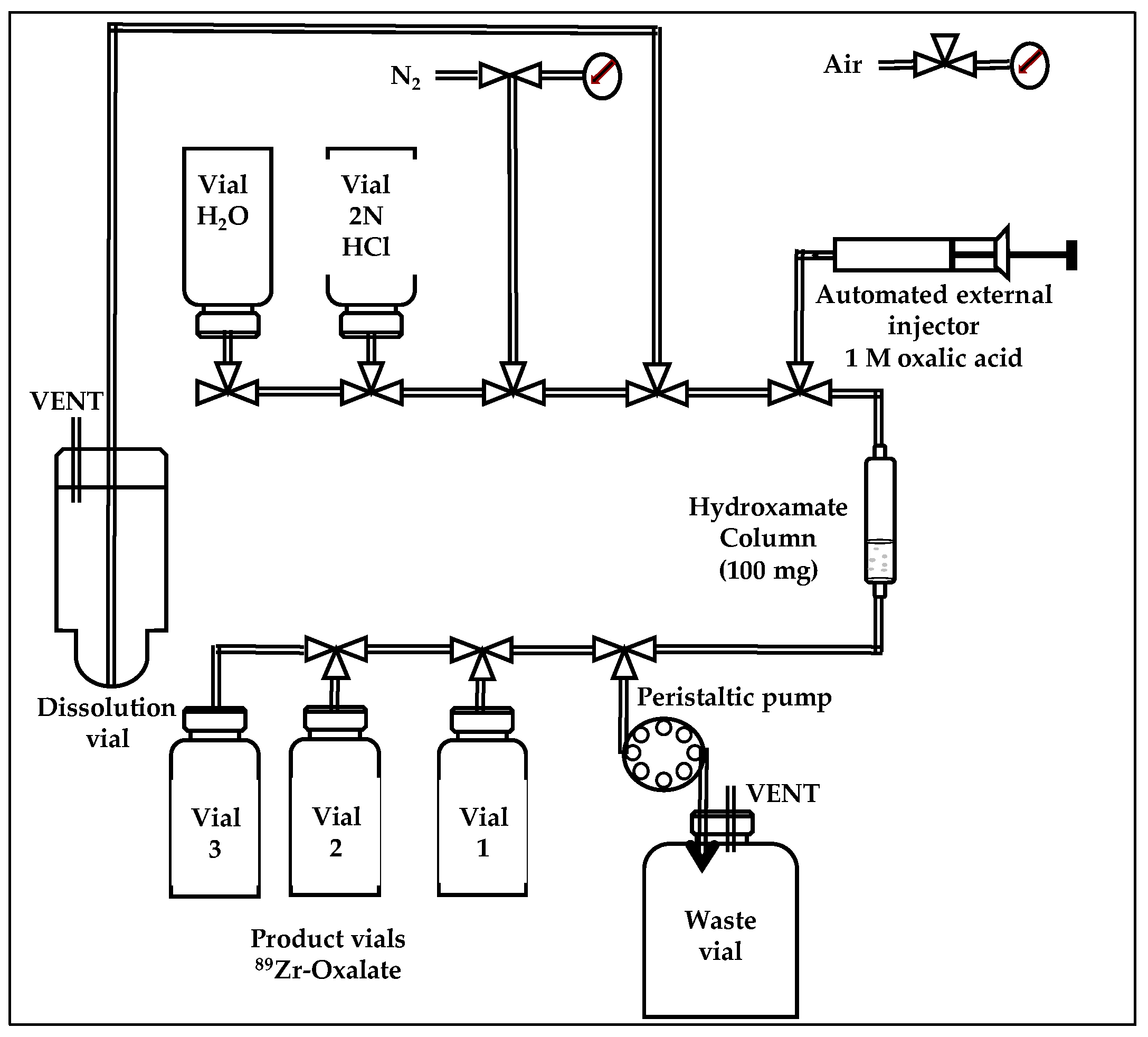
2.4. Automated Cassette-Based Module and Separation Chemistry for 89Zr-oxalate
A cassette-based purification module obtained from ACSI was modified in-house to remotely produce
89Zr-oxalate inside a hot-cell. The front panel of the module was equipped with two cassettes, one to allow for easy load of the liquid vials, the other for dispensing of three vials (
Figure 2 and
Figure S2).
The glass dissolution vial was prewashed with concentrated 6N trace metal HCl for at least 24 h. Prior to perform purification, three and five manifold kits were installed and the unit was cleaned using 6N trace metal HCl. To avoid solvent leaks, all the liquids were transferred through the system by polytetrafluoroethylene (PTFE) tubing while using a negative pressure generated by a peristaltic pump with a flow rate of 4 mL/min. To reduce exposure to metal that can affect EMA, all the liquid and gas connections were assembled while using PTFE and silicon connectors. Before each production, all solutions (30 mL of 2N trace metal HCl, 20 mL of high-purity water, and 3 mL of 1 M oxalic acid) were preloaded into the module. Hydroxamate resin (100 mg) prepared and functionalized following the procedure reported by Holland et al. was packed in a 1 mL cartridge between two frits (20 microns) (United chemical technologies, Bristol, PA, USA) [
5]. The hydroxamate column was preconditioned with 8 mL of acetonitrile, 20 mL of high-purity water, and 3 mL of 2N trace metal HCl. The module was controlled by a laptop computer and the module functions by an easy to program user interface (RSView 32 software, Rockwell Software, Milwaukee, WI, USA).
After irradiation and decay of the short half-life 89mZr (t1/2 = 4.16 min), either 89Y/89Zr foil, or pressed target was removed from the degrader/holder and was directly dropped in the dissolution vial and then dissolved with 10 mL of 2N trace metal HCl for 3–5 min at room temperature. If the foil and pressed targets overheated, they required more time for a complete dissolution in HCl. The crude solution was then loaded and passed through the hydroxamate resin in the cartridge and transferred to the waste using a negative pressure created by the peristaltic pump. The column was washed with 2N trace metal HCl (20 mL) and high-purity water (20 mL). Then, a low helium pressure (20 psi) was applied to dry the line and the cartridge. Finally, the Zr-89 bound to the column was eluted with 0.5 mL of 1M oxalic acid under helium pressure (20 psi). This process was repeated twice to collect three 0.5-mL fractions in different vials. EMA was determined for each fraction. The fourth fraction was collected for half-life measurement.
2.5. Effective Molar Activity Using DFO Chelator
EMA (GBq/µmol) of 89Zr was calculated via titration with DFO and the purified combined 89Zr-oxalate fractions 1 and 2. Solutions of DFO (0.5 mL in 1.5-mL microcentrifuge tube) at different concentrations (380.6 to 2.3 × 10-5 nmol) were prepared via serial dilution. After 1 h incubation time with 89Zr (0.9 to 1.1 MBq, 50 μL), the solutions of DFO were quenched while using DTPA (40 μL, 1 mmol) for 10 min to complex free 89Zr. After incubation and quenching with DTPA, the EMA was determined by measuring the DFO labelling efficiency with ITLC-SG analysis using DTPA (100 mM) as a mobile phase solvent. 89Zr-DTPA complex eluted with the solvent front, whilst 89Zr-DFO complex remained at the origin. ITLC-SG was analyzed using Radio-ITLC scanner. The binding percentages were plotted on a sigmoidal dose-response curve. EC50 value, which is reflecting the molar concentration at 50%, was calculated by prism 7 software (GraphPad Software Inc, La Jolla, CA, USA). The EMA was calculated as two times the EC50 value.
2.6. Determination of Radionuclide and Metal Impurities
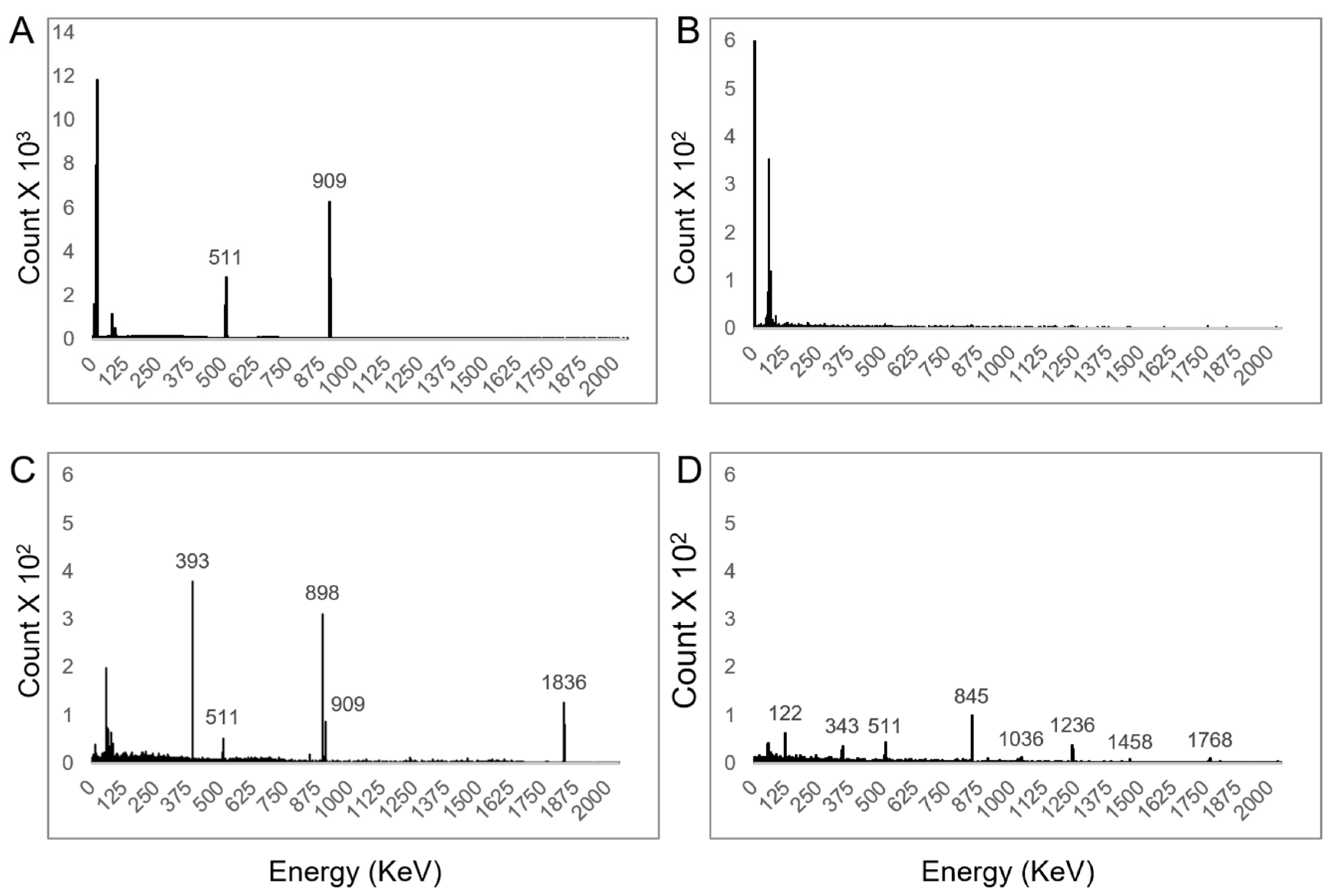
Samples containing 148–222 kBq of purified fraction of 89Zr-oxalate in a 1.5 mL microcentrifuge tube were diluted with high-purity water to bring a final volume of 0.5 mL. The radionuclidic purity was determined by γ–ray spectroscopy on a HPGe detector with zoom energy window of 1–2000 KeV. With that amount of radioactivity, the dead time was below 5%. Samples were counted for only 10 min after the end of synthesis. In addition, the test was repeated after six months to qualify radionuclide impurities with long half-life, such as 88Y (t1/2 = 106.6 d) and 88Zr (t1/2 = 83.4 d); they were not present in the formulated 89Zr. The half-life was estimated using dose calibrator CRC-55 PET and was calibrated at 465. Trace metal analysis in 89Zr-oxalate solutions that were produced while using 89Y-foil and 89Y-pressed powder as starting materials was performed by inductively coupled plasma mass spectroscopy (ICP-MS) for 31 metals in compliance with USP <233> (Exova, St-Augustin-de-Desmaures, QC, Canada).
4. Discussion
According to data reported by Mustafa et al., the optimal cross-section is at 750.8 ± 1.5 mb while using 12.46 MeV
Ep [
7]. Increasing
Ep beyond this level will increase the
89Zr production yield, but long half-life by-products, such
88Zr and
88Y, will then be produced. Thus,
89Zr production is not optimal using medium energy cyclotron, except considering the reduction of the
Ep, which can be achieved with an energy degrader. We designed different size target degraders and holders in different materials (
Figure 1) to be used with incident
Ep of 16.2–18 MeV. An aluminum holder/degrader was first selected for its high-energy transfer [
13], which renders more efficient target cooling during the irradiation. Niobium has high chemical resistance and was selected due to its high melting temperature and to avoid any deterioration of the target body during cleaning with hydrochloric acid to remove any trace of unknown metals [
14].
The degradation of the beam energy with our designed degraders was achieved without damage, except for the Nb degrader while using long irradiation times (
Figure 1B). Under these conditions, the
89Y-pressed powder oxidized into Y
2O
3, which is poorly soluble and required a high volume of 12N HCl for its dissolution and purification [
24]. Furthermore, the aluminum degrader presents better heat transfer and no local overheating was observed for the Al-21-1 degrader (
Figure 1A) when 15 and 40 μA were applied to
89Y-foil at 13.3 and 15 MeV for 4.5 and 1.5 h, respectively.
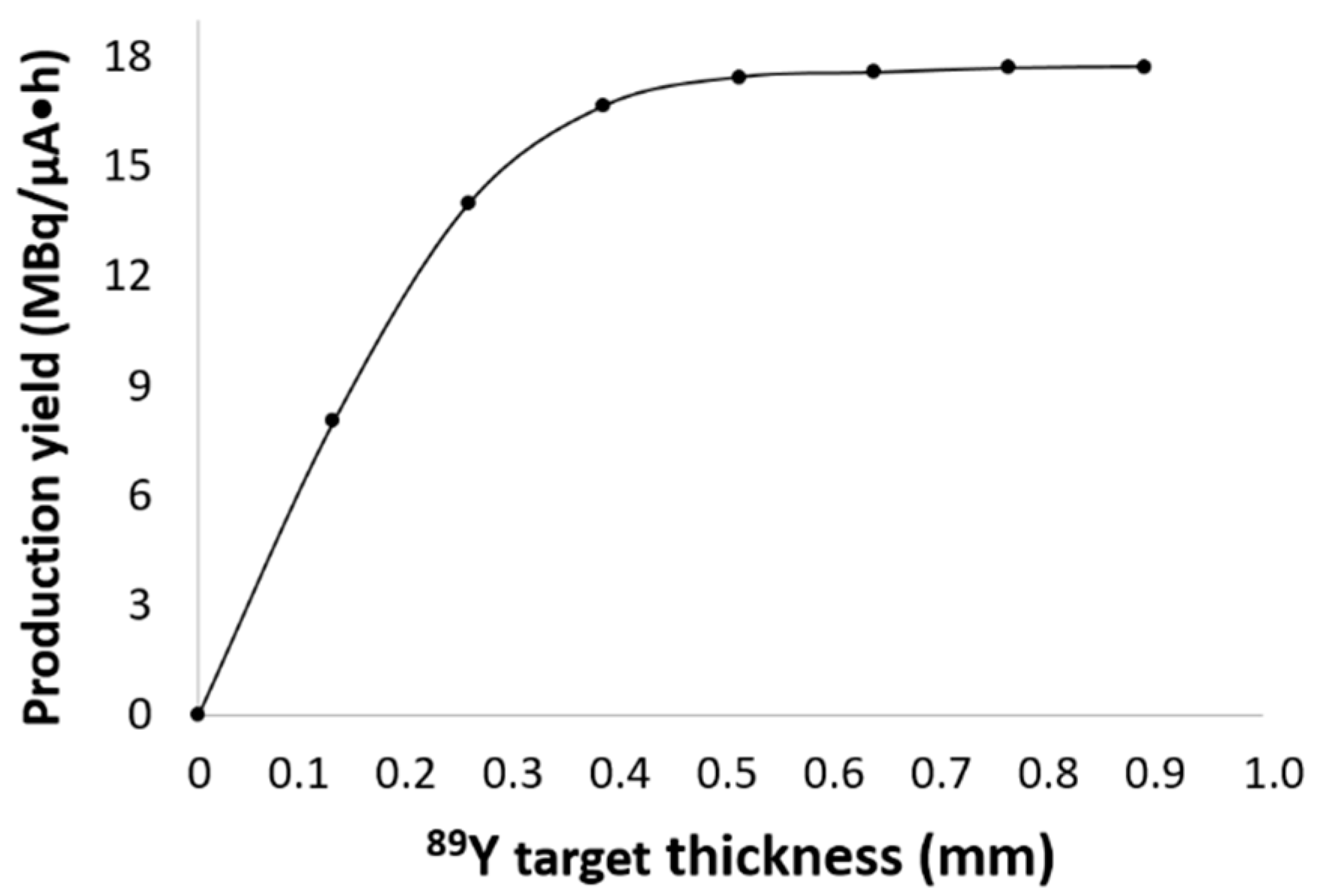
The production yield depends on the incident
Ep, the degrader material, and the beam size, as well as the density and thickness of the target materials. The highest yields were found for 0.50-mm thick
89Y-foil target (two foils). Increasing the number of foils beyond this thickness did not improve the yield for an
Ep of 11.3 MeV. The production yield was also affected by the target diameter. Indeed, higher yields were obtained with the 13-mm diameter targets that provided a better fit with the beam size of ~10 mm. One should note that a current as high as 40 μA could be applied when the Al-13 holder was used as compared to all other degraders, the cooling system being more efficient. Indeed, the Al back support is thinner for the Al-13 holder (0.5 mm) as compared to that of other degraders (1.0 mm) (
Figure 1). In addition, high production yield values were obtained while using both foil and pressed targets.
The highest yield (54 ± 2.9 MBq/μA·h) obtained at 15.0 MeV is similar to that the one reported by Holland et al. (56.24 ± 4.1 MbBq/μA·h) [
5] for a similar foil target thickness. As shown in
Table 2, the production yield reported for
89Y-pressed target (49.5 ± 1.2 MBq/μA·h) is slightly lower than the one that was reported by Zweit et al. (66.6 MBq/μA·h) while using
89Y-oxide pressed targets irradiated at 16.2 MeV from deuteron [
8]. Zweit et al. reported that the increase of beam current and irradiation time was more challenging using pressed targets [
8]. In the present study, we were able to irradiate at 15 and 40 μA for 4.2 and 1.5 h, respectively, without overheating the
89Y-target material.
The automated cassette-based module that was used in this work is simple, reliable, and provides reproducible results with high-recovered activity. The system can accommodate three separate product vials with independent controlled valves that gives an advantage over the automated purification box developed by Wooten et al. that eluted purified
89Zr in only one fraction vial [
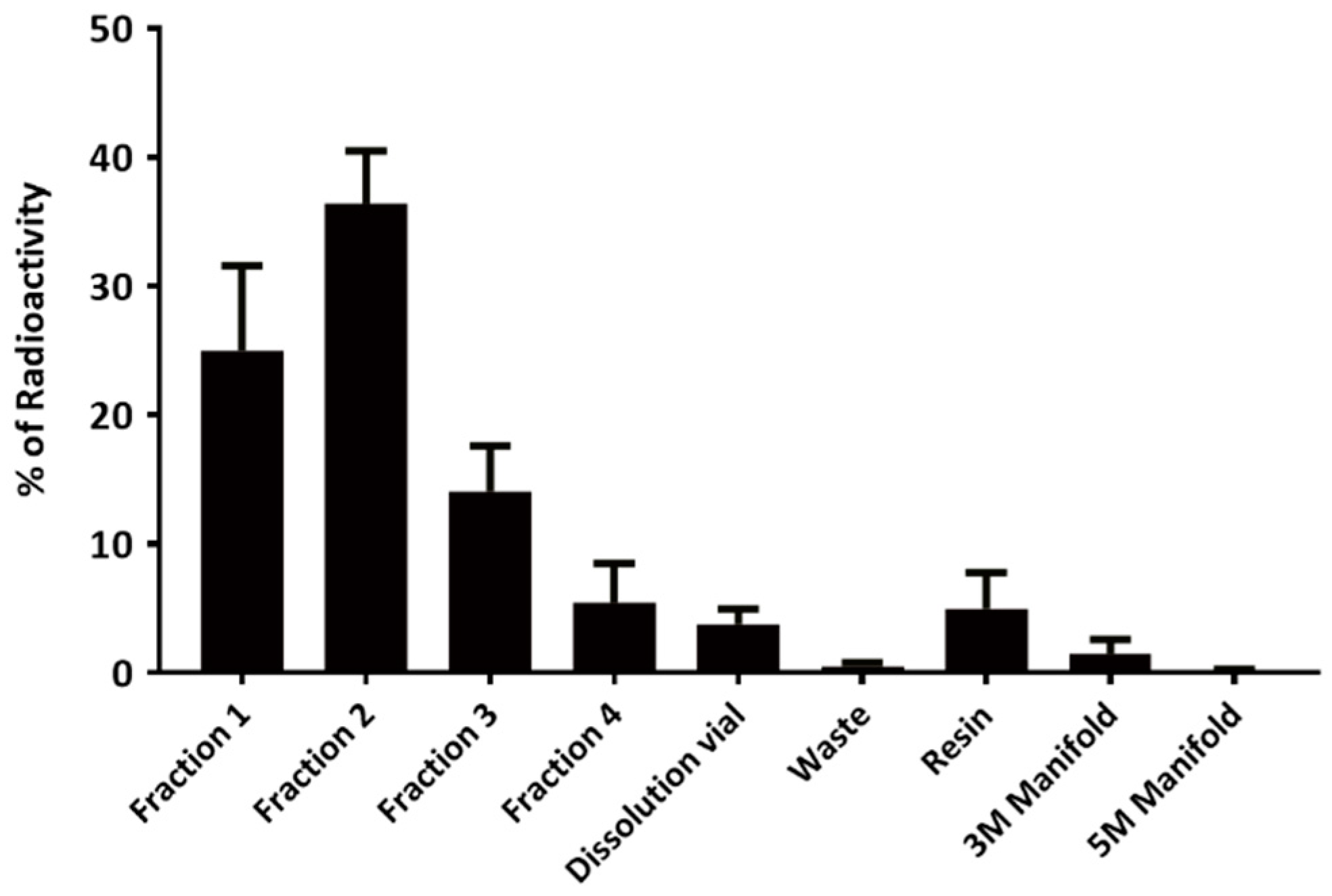
14]. As the maximum activity and EMA were found in fractions 1 and 2 (
Figure 4 and
Table 3), we usually prefer to combine only these two fractions for further studies. Using our system, greater EMA (1378 to 8892 vs. 5 to 353 Ci/mmol), more efficient separation with higher
89Zr recovered activity (81 ± 4.4% vs. 74 ± 16%) and lower
89Zr retention on the column (5 ± 2.8% vs. 18 ± 15%) were obtained, when compared to Wooten’s results [
14].
Commonly, to achieve high EMA, the chemical must be metal trace with high purity
89Y. To our knowledge, we reported in these studies the highest EMA values for
89Zr that were produced by cyclotron by optimizing the
Ep, the amount/thickness and the current applied on the
89Y-target. The highest EMA values were obtained while using packed
89Y-foils. The EMA values achieved while using
89Y-pressed targets well improves the value reported by Holland et al. [
5]. Pressed
89Y targets are easy to prepare compared to electro-deposition preparation methods, which are also time consuming and less expensive compared to
89Y-foil. They could represent an excellent alternative for routine production of
89Zr. The preparation of both foil and pressed targets can be done in a few minutes. The cost for
89Y-foil is ~108 US
$/irradiation using 2 foils (0.5-mm thickness and 13-mm diameter), while for
89Y powder (0.5-mm thickness, 13-mm diameter, and 250 mg), the cost is ~3 US
$/irradiation, without considering the equipment costs. Admittedly, the use of pressed target comes at the cost of purchasing a press. This alternative strategy can rapidly be cost-effective if the press is used in the development of other solid targets and/or if the price of
89Y-foil rises.
One should note that
89Y-powder 40-mesh (99.6% purity) that was used in this work contains more impurities than
89Y-foil (99.9% purity). Particularly, 2000 ppm of Fe
+3 in
89Y powder is responsible for the presence of the low amount of long half-life radiometal impurities (
56,57,58Co) observed six months post irradiation. However, the metal impurity contents were almost similar for purified
89Zr-oxalate solutions obtained from
89Y-foil and pressed targets and it did not affect the labelling of
89Zr with deferoxamine chelator (
Table 4).
Using our irradiation parameters and 11.3 MeV Ep for production of 89Zr, no radionuclide impurities, such as 88Zr and 88Y were observed in purified fractions. However, when we increased the Ep to 13.3 MeV, trace of 88Zr and 88Y were observed on γ-ray spectrum measured after six months in the residual 89Zr product solution after decay. The presence of 88Y in the product solution after 89Zr decay is not associated with the poor separation efficiency of the hydroxamate column, but is related to the decay chain of 88Zr to 88Y. Ep increases the production yield as well as the EMA, but it also increases production of long half-life impurities. This contamination will not affect image quality nor increase significantly the radiation exposure of the patient, but it must be considered when managing radioactive waste.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}