Analysis of the Molecular Interactions between Cytochromes P450 3A4 and 1A2 and Aflatoxins: A Docking Study


 , , , and
, , , and 
Abstract
1. Introduction
2. Computational Details
2.1. Quantum Chemical Calculations
2.2. Preparation of Proteins for Molecular Docking Calculations
2.3. Molecular Docking
2.4. Chemical Reactivity and Energy Calculations
3. Results and Discussion
3.1. Geometry Optimization
3.2. Docking Analysis
3.2.1. Molecular Interactions between CYP 3A4 (1TQN PDB ID) and AFB1, B2, G1, and G2
3.2.2. Molecular Interactions between CYP1A2 (2HI4 PDB ID) and AFB1, B2, G1, and G2
3.2.3. Molecular Interactions of CYP3A4 with AFB1 Using Different PDB IDs
3.3. Quantum Chemical Calculations
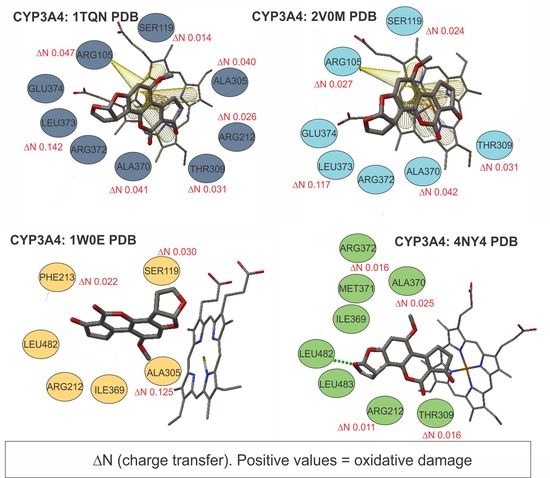
3.3.1. Chemical Reactivity and Charge Transfer Calculations (ΔN) for Protein CYP3A4 (1TQN, 20VM, 4NY4, and 1W0E PDB) with AFB1
3.3.2. Chemical Reactivity and Charge Transfer Calculations (ΔN) for Protein CYP1A2 (2HI4 PDB) with AFB1
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bbosa, G.S.; Kitya, D.; Lubega, A.; Ogwal-Okeng, J.; Anokbonggo, W.W.; Kyegombe, D.B. Review of the Biological and Health Effects of Aflatoxins on Body Organs and Body Systems. In Aflatoxins-Recent Advances and Future Prospects; Razzaghi-Abyaneh, M., Ed.; InTech: Rijeka, Croatia, 2013; Ch. 12. [Google Scholar] [CrossRef]
- Sweeney, M.J.; Dobson, A.D.W. Mycotoxin production by Aspergillus, Fusarium and Penicilliium species. Int. J. Food Microbiol. 1998, 43, 141–158. [Google Scholar] [CrossRef]
- Lizárraga-Paulín, E.G.; Moreno-Martínez, E.; Miranda-Castro, S.P. Aflatoxins and Their Impact on Human and Animal Health: An Emerging Problem. In Aflatoxins—Biochemistry and Molecular Biology; InTech: Rijeka, Croatia, 2011; pp. 255–284. [Google Scholar]
- Anttila, A.; Bhat, R.V.; Bond, J.A.; Borghoff, S.J.; Bosch, F.X.; Carlson, G.P.; Castegnaro, M.; Cruzan, G.; Gelderblom, W.C.; Hass, U.; et al. IARC monographs on the evaluation of carcinogenic risks to humans. Some traditional herbal medicines, some mycotoxins, naphthalene and styrene. IARC Monogr. Eval. Carcinog. Risks Hum. 2002, 82, 1–556. [Google Scholar] [CrossRef]
- Kensler, T.W.; Roebuck, B.D.; Wogan, G.N.; Groopman, J.D. Aflatoxin: A 50-year Odyssey of mechanistic and translational toxicology. Toxicol. Sci. 2011, 120 (Suppl. 1), 28–48. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Moreno, M. Metabolic Changes of Aflatoxin B1 to become an Active Carcinogen and the Control of this Toxin. Immunome Res. 2015, 11, 1. [Google Scholar] [CrossRef]
- González, J.C.R.; Guerra, I.R. El sistema citocromo P450 y el metabolismo de xenobióticos Cytochrome P450 system and xenobiotic metabolism. Rev. Cuba. Farm. 2014, 48, 495–507. [Google Scholar]
- Guengerich, F.P.; Waterman, M.R.; Egli, M. Recent Structural Insights into Cytochrome P450 Function. Trends Pharmacol. Sci. 2016, 37, 625–640. [Google Scholar] [CrossRef]
- Landeros-Martinez, L.-L.; Flores-Holguín, N. Predicción de la reactividad química de tamoxifeno en receptores hormonales. Mem. Congr. Investig. Cient. Multidiscip. 2017, 4, 24–38. [Google Scholar]
- Jett, J.E.; Lederman, D.; Wollenberg, L.A.; Li, D.; Flora, D.R.; Bostick, C.D.; Tracy, T.S.; Gannett, P.M. Measurement of electron transfer through cytochrome p450 protein on nanopillars and the effect of bound substrates. J. Am. Chem. Soc. 2013, 135, 3834–3840. [Google Scholar] [CrossRef][Green Version]
- Kumar, V.; Jain, G.; Kishor, S.; Ramaniah, L.M. Chemical reactivity analysis of some alkylating drug molecules—A density functional theory approach. Comput. Theor. Chem. 2011, 968, 18–25. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other function. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Amovilli, C.; Barone, V.; Cammi, R.; Cancès, E.; Cossi, M.; Mennucci, B.; Pomelli, C.S.; Tomasi, J. Recent Advances in the Description of Solvent Effects with the Polarizable Continuum Model. In Advances in Quantum Chemistry; Löwdin, P.-O., Ed.; Academic Press: Cambridge, MA, USA, 1998; Volume 32, pp. 227–261. [Google Scholar]
- Mennucci, B.; Cancès, E.; Tomasi, J. Evaluation of Solvent Effects in Isotropic and Anisotropic Dielectrics and in Ionic Solutions with a Unified Integral Equation Method: Theoretical Bases, Computational Implementation, and Numerical Applications. J. Phys. Chem. B 1997, 101, 10506–10517. [Google Scholar] [CrossRef]
- Van Soest, T.C.; Peerdeman, A.F. The crystal structures of aflatoxin B1. I. The structure of the chloroform solvate of aflatoxin B1 and the absolute configuration of aflatoxin B1. Acta Cryst. B 1970, 26, 1940–1947. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Yano, J.K.; Wester, M.R.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05 A resolution. J. Biol. Chem. 2004, 279, 38091–38094. [Google Scholar] [CrossRef]
- Williams, P.A.; Cosme, J.; Vinković, D.M.; Ward, A.; Angove, H.C.; Day, P.J.; Vonrhein, C.; Tickle, I.J.; Jhoti, H. Crystal Structures of Human Cytochrome P450 3A4 Bound to Metyrapone and Progesterone. Science 2004, 305, 683–686. [Google Scholar] [CrossRef]
- Brändén, G.; Sjögren, T.; Schnecke, V.; Xue, Y. Structure-based ligand design to overcome CYP inhibition in drug discovery projects. Drug Discov. Today 2014, 19, 905–911. [Google Scholar] [CrossRef]
- Ekroos, M.; Sjögren, T. Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc. Natl. Acad. Sci. USA 2006, 103, 13682–13687. [Google Scholar] [CrossRef]
- Sansen, S.; Yano, J.K.; Reynald, R.L.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Adaptations for the oxidation of polycyclic aromatic hydrocarbons exhibited by the structure of human P450 1A2. J. Biol. Chem. 2007, 282, 14348–14355. [Google Scholar] [CrossRef]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific LLC: San Carlos, CA, USA, 2002. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Martinez-Araya, J.I.; Glossman-Mitnik, D. Assessment of ten density functionals through the use of local hyper—Softness to get insights about the catalytic activity Iron—Based organometallic compounds for ethylene polymerization as testing molecules. J. Mol. Model. 2018, 24, 42. [Google Scholar] [CrossRef] [PubMed]
- Bonomo, S.; Jørgensen, F.S.; Olsen, L. Dissecting the Cytochrome P450 1A2- and 3A4-Mediated Metabolism of Aflatoxin B1 in Ligand and Protein Contributions. Chem. Eur. J. 2017, 23, 2884–2893. [Google Scholar] [CrossRef] [PubMed]
- Sohl, C.D.; Isin, E.M.; Eoff, R.L.; Marsch, G.A.; Stec, D.F.; Guengerich, F.P. Cooperativity in oxidation reactions catalyzed by cytochrome P450 1A2: Highly cooperative pyrene hydroxylation and multiphasic kinetics of ligand binding. J. Biol. Chem. 2008, 283, 7293–7308. [Google Scholar] [CrossRef] [PubMed]
- Bren, U.; Fuchs, J.E.; Oostenbrink, C. Cooperative binding of aflatoxin B1 by cytochrome P450 3A4: A computational study. Chem. Res. Toxicol. 2014, 27, 2136–2147. [Google Scholar] [CrossRef] [PubMed]
- Sevrioukova, I.F.; Poulos, T.L. Understanding the mechanism of cytochrome P450 3A4: Recent advances and remaining problems. Dalton Trans. 2013, 42, 3116–3126. [Google Scholar] [CrossRef]
- Gay, S.C.; Roberts, A.G.; Halpert, J.R. Structural Features of Cytochromes P450 and Ligands that Affect Drug Metabolism as Revealed by X-ray Crystallography and NMR. Future Med. Chem. 2010, 2, 1451–1468. [Google Scholar] [CrossRef]
- Wu, J.; Chen, R.; Zhang, C.; Li, K.; Xu, W.; Wang, L.; Chen, Q.; Mu, P.; Jiang, J.; Wen, J.; et al. Bioactivation and regioselectivity of pig cytochrome P450 3A29 towards aflatoxin B1. Toxins 2016, 8, 267. [Google Scholar] [CrossRef]
- Wang, X.; Song, L.; Tian, C.; He, J.; Wang, S.; Wang, J.; Li, C. DFT Investigation of the Effects of Coexisting Cations and Complexing Reagents on Ni(II) Adsorption by a Polyvinylidene Fluoride-Type Chelating Membrane Bearing Poly(Amino Phosphonic Acid) Groups. Metals 2017, 7, 61. [Google Scholar] [CrossRef]
- Song, L.Z.; Zhou, W.Y.; Wang, X.L.; He, J.; Han, K.D.; Niu, L.J. Interaction of Chlorpyrifos with Purine Bases: A Study of Electrochemical Measurement and Density Functional Theory Calculation. Asian J. Chem. 2015, 27, 2445–2451. [Google Scholar] [CrossRef]
- Dutkiewicz, Z.; Mikstacka, R. Structure-Based Drug Design for Cytochrome P450 Family 1 Inhibitors. Bioinorg. Chem. Appl. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Legnani, L.; Compostella, F.; Sansone, F.; Toma, L. Cone Calix[4]arenes with Orientable Glycosylthioureido Groups at the Upper Rim: An In-Depth Analysis of Their Symmetry Properties. J. Org. Chem. 2015, 80, 7412–7418. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | DFT Functional/Basis set 6-311G(d,p) | Experimental Reference (a) | |||
|---|---|---|---|---|---|
| Gas | Solvent (water) | ||||
| B3LYP | M06-2X | B3LYP | M06-2X | ||
| Bond Distance (Å) | |||||
| 20C-22C | 1.32 | 1.32 | 1.32 | 1.32 | 1.30 |
| 20C-12C | 1.51 | 1.51 | 1.51 | 1.51 | 1.50 |
| 12C-15C | 1.56 | 1.55 | 1.55 | 1.55 | 1.55 |
| 15C-3O | 1.41 | 1.40 | 1.41 | 1.40 | 1.41 |
| 3O-22C | 1.38 | 1.37 | 1.38 | 1.37 | 1.38 |
| 22C-31H | 1.07 | 1.07 | 1.07 | 1.07 | 1.04 |
| 20C-28H | 1.07 | 1.07 | 1.07 | 1.07 | 1.06 |
| 12C-23H | 1.09 | 1.09 | 1.09 | 1.09 | 1.01 |
| Correlation (r) | 0.9917 | 0.9907 | 0.9926 | 0.9918 | |
| Bond Angles (°) | |||||
| 22C-3O-15C | 107.5 | 107.7 | 107.5 | 107.7 | 107.2 |
| 3O-15C-12C | 107.5 | 107.4 | 107.5 | 107.4 | 107.5 |
| 15C-12C-20C | 101.0 | 101.0 | 101.0 | 101.0 | 101.0 |
| 31H-22C-20C | 130.8 | 130.6 | 130.9 | 130.7 | 132.0 |
| 28H-20C-22C | 126.9 | 127.1 | 124.7 | 126.6 | 129.0 |
| 31H-22C-3O | 114.0 | 114.2 | 114.2 | 114.4 | 113.0 |
| 28H-2OC-12C | 124.5 | 124.5 | 124.7 | 124.8 | 122.0 |
| 23H-12C-15C | 113.1 | 113.5 | 112.7 | 113.0 | 114.0 |
| 23H-12C-20C | 112.5 | 112.9 | 112.7 | 113.1 | 113.0 |
| Correlation (r) | 0.9708 | 0.9751 | 0.9525 | 0.9688 | |
| Ligand | DFT Functional/ 6-311G(d,p) | Binding Energy (kcal/mol) | Active Site | Hydrogen Bonds | π–π Interactions |
|---|---|---|---|---|---|
| AFB1 | B3LYP | −8.53 | Arg105, Ser119, Arg212, Ala305, Thr309, Ala370, Arg372, Leu373, Glu374, HEM508 | 0 | 5 |
| M06-2X | −8.78 | Arg105, Ser119, Arg212, Ala305, Thr309, Ala370, Arg372, Leu373, Glu374, HEM508 | 0 | 5 | |
| AFG1 | B3LYP | −8.44 | Arg105, Ser119, Arg212, Ala305, Ala370, Arg372, Leu373, Glu374, HEM 508 | Arg212 – O5 Ser119 – O7 | 2 |
| M06-2X | −8.70 | Arg105, Ser119, Arg212, Ala305, Ala370, Arg372, Leu373, Glu374, HEM 508 | Arg212 – O5 Ser119 – O7 | 2 | |
| AFB2 | B3LYP | −8.77 | Arg105, Ser119, Arg212, Ala305, Thr309, Ala370, Arg372, Leu373, Glu374, HEM508 | 0 | 5 |
| M06-2X | −8.86 | Arg105, Ser119, Arg212, Ala305, Thr309, Ala370, Arg372, Leu373, Glu374, HEM508. | 0 | 5 | |
| AFG2 | B3LYP | −8.40 | Arg105, Ser119, Arg212, Ala305, Ala370, Arg372, Leu373, Glu374, HEM508 | Ser119 – O7 Arg212 – O5 | 2 |
| M06-2X | −8.39 | Arg105, Ser119, Arg212, Ala305, Ala370, Arg372, Glu374, HEM508 | Ser119 – O7 Arg212 – O5 | 2 |
| Ligand | DFT Functional/ 6-311G(d,p) | Binding Energy (kcal/mol) | Active Site | Hydrogen Bonds | π–π Interactions |
|---|---|---|---|---|---|
| AFB1 | B3LYP | −6.66 | Thr124, Phe125, Phe226, Asp313, Gly316, Ala317 Asp320, Thr321, Leu382, Ile386, Thr498, HEM900 | 0 | 0 |
| M06-2X | −6.96 | Thr124, Phe125, Phe226, Asp313, Gly316, Ala317, Asp320, Thr321, Leu382, Ile386, HEM 900 | 0 | 0 | |
| AFB2 | B3LYP | −8.41 | Ile117, Thr118, Thr124, Phe125, Phe226, Val227, Asn312, Asp313, Ala317, Thr321, HEM900 | 0 | 0 |
| M06-2X | −6.35 | Thr124, Phe125, Phe226, Asp313, Gly316, Ala317, Asp320, Trh321, Leu382, Ile386, Thr498, HEM900 | Asp320 –O4 | 0 | |
| AFG1 | B3LYP | −4.92 | Thr124, Phe125, Phe226, Asp313, Gly316, Ala317, Asp320, Thr321, Leu382, Ile386, Thr498, HEM900 | Gly316-O4 | 0 |
| M06-2X | −5.52 | Thr124, Phe125, Asp313, Gly316, Ala317, Asp320, Thr321, Leu382, Ile386, Leu497, Thr498, HEM900 | 0 | 0 | |
| AFG2 | B3LYP | −3.51 | Thr124, Phe125, Phe226, Asp313, Gly316, Ala317, Asp320, Thr321, Leu382, Ile386, Leu497, Thr498, HEM900 | 0 | 0 |
| M06-2X | −3.97 | Thr124, Phe125, Phe226, Asp313, Gly316, Ala317, Asp320, Thr321, Leu382, Ile386, Leu497, Thr498, HEM900 | 0 | 0 |
| 1TQN | 2V0M | 1W0E | 4NY4 | |
|---|---|---|---|---|
| Active Site | Arg105, Ser119, Arg212, Ala305, Thr309, Ala370, Arg372, Leu373, Glu374, HEM508 | Arg105, Ser119, Thr309, Ala370, Arg372, Leu373, Glu374, HEM497 | Ser119, Arg212, Phe213, Ala305, Ile369, Leu482, HEM501 | Arg212, Thr309, Ile369, Ala370, Met371, Arg372, Leu482, Leu483, HEM601 |
| Residue | PDB | EHOMO (eV) | ELUMO (eV) | Electron Affinity (eV) | Ionization Potential (eV) | Hardness (eV) | Chemical Potential (eV) |
|---|---|---|---|---|---|---|---|
| AFB1 | 1TQN | −7.4236 | −1.1978 | 1.1978 | 7.4236 | 3.1128 | −4.3107 |
| 1W0E | −6.6317 | −2.0710 | 2.0710 | 6.6317 | 2.2803 | −4.3514 | |
| 20VM | −6.7898 | −2.1992 | 2.1992 | 6.7898 | 2.2953 | −4.4945 | |
| 4NY4 | −6.5922 | −2.0506 | 2.0506 | 6.5922 | 2.2708 | −4.3214 | |
| ARG105 | 1TQN | −7.1661 | −0.7281 | 0.7281 | 7.1661 | 3.2190 | −3.9471 |
| 1W0E | Not present | ||||||
| 20VM | −7.3185 | −1.0767 | 1.0767 | 7.3185 | 3.1209 | −4.1976 | |
| 4NY4 | Not present | ||||||
| SER119 | 1TQN | −7.4236 | −1.1978 | 1.1978 | 7.4236 | 3.1128 | −4.3107 |
| 1W0E | −7.1000 | −0.9390 | 0.9390 | 7.1000 | 3.0804 | −4.0195 | |
| 20VM | −7.5033 | −0.9355 | 0.9355 | 7.5033 | 3.2839 | −4.2194 | |
| 4NY4 | Not present | ||||||
| ARG212 | 1TQN | −7.6189 | −0.7007 | 0.7007 | 7.6189 | 3.4591 | −4.1598 |
| 1W0E | Not present | ||||||
| 20VM | Not present | ||||||
| 4NY4 | −7.5411 | −0.8419 | 0.8419 | 7.5411 | 3.3496 | −4.1915 | |
| ARG212-PHE213 | 1TQN | Not present | |||||
| 1W0E | −7.2195 | −1.0068 | 1.0068 | 7.2195 | 3.1063 | −4.1131 | |
| 20VM | Not present | ||||||
| 4NY4 | Not present | ||||||
| THR309 | 1TQN | −7.2559 | −0.9943 | 0.9943 | 7.2559 | 3.1308 | −4.1251 |
| 1W0E | Not present | ||||||
| 20VM | −7.1833 | −1.1320 | 1.1320 | 7.1833 | 3.0256 | −4.1576 | |
| 4NY4 | −7.1795 | −1.1137 | 1.1137 | 7.1795 | 3.0328 | −4.1466 | |
| ALA305 | 1TQN | −7.1635 | −0.8966 | 0.8966 | 7.1634 | 3.1334 | −4.0300 |
| 1W0E | −7.2578 | 0.8408 | −0.8408 | 7.2578 | 4.0493 | −3.2085 | |
| 20VM | Not present | ||||||
| 4NY4 | Not present | ||||||
| ALA370 | 1TQN | −7.1631 | −0.8740 | 0.8740 | 7.1631 | 3.1445 | −4.0186 |
| 1W0E | Not present | ||||||
| 20VM | −7.2899 | −0.7562 | 0.7562 | 7.2899 | 3.2668 | −4.0231 | |
| 4NY4 | −7.1765 | −0.9069 | 0.9069 | 7.1765 | 3.1347 | −4.0417 | |
| ARG372 | 1TQN | Not present | |||||
| 1W0E | Not present | ||||||
| 20VM | Not present | ||||||
| 4NY4 | −7.4102 | −0.8628 | 0.8628 | 7.4102 | 3.2736 | −4.1365 | |
| ARG372-LEU373-GLU374 | 1TQN | −5.8205 | −0.3096 | 0.3096 | 5.8205 | 2.7554 | −3.0651 |
| 1W0E | Not present | ||||||
| 20VM | −5.8007 | −0.9638 | 0.9638 | 5.8007 | 2.4184 | −3.3822 | |
| 4NY4 | Not present | ||||||
| HEM | 1TQN | −0.6215 | 0.4008 | −0.4008 | 0.6215 | 0.5111 | −0.1103 |
| 1W0E | −0.0157 | 0.6718 | −0.6718 | 0.0157 | 0.3438 | 0.3280 | |
| 20VM | −0.2140 | 0.8375 | −0.8375 | 0.2144 | 0.5260 | 0.3115 | |
| 4NY4 | −0.9382 | 0.4794 | −0.4794 | 0.9382 | 0.7088 | −0.2293 | |
| Residue/PDB | 1TQN | 1W0E | 20VM | 4NY4 |
|---|---|---|---|---|
| ARG105 | 0.048 | -- | 0.027 | -- |
| SER119 | 0.014 | 0.031 | 0.025 | -- |
| ARG212 | 0.027 | -- | -- | 0.012 |
| ARG212-PHE213 | -- | 0.22 | -- | -- |
| THR309 | 0.032 | -- | 0.032 | 0.016 |
| ALA305 | 0.041 | 0.126 | -- | -- |
| ALA370 | 0.042 | -- | 0.042 | 0.026 |
| ARG372-LEU373-GLU374 | 0.142 | -- | 0.118 | -- |
| ARG372 | -- | -- | -- | 0.017 |
| HEM | 0.816 | 0.892 | 0.852 | 0.687 |
| Residue | EHOMO (ev) | ELUMO (ev) | Electron Affinity (ev) | Ionization Potential (ev) | Hardness (ev) | Chemical Potential (ev) |
|---|---|---|---|---|---|---|
| AFB1 | −6.7195 | −2.2245 | 2.2245 | 6.7195 | 2.2787 | −4.5032 |
| THR124- PHE125 | −7.0529 | −1.2218 | 1.2218 | 7.0529 | 2.9155 | −4.1373 |
| ASP313 | −9.7003 | −8.7071 | 8.7071 | 9.7003 | 0.4966 | −9.2037 |
| PHE226 | −7.0339 | −0.7202 | 0.7202 | 7.0339 | 3.1568 | −3.8771 |
| GLY316- ALA317 | −7.0088 | −0.8041 | 0.8041 | 7.0088 | 3.1023 | −3.9065 |
| ASP321- THR322 | −8.9063 | −8.0998 | 8.0998 | 8.9063 | 0.4032 | −8.5030 |
| LEU382 | −7.2668 | −0.5047 | 0.5047 | 7.2668 | 3.3810 | −3.8858 |
| ILE386 | −6.9087 | −0.9583 | 0.9583 | 6.9087 | 2.9751 | −3.9335 |
| THR498 | −7.2769 | −0.7741 | 0.7741 | 7.2769 | 3.2513 | −4.0255 |
| HEM900 | −1.0552 | 0.5227 | −0.5227 | 1.0552 | 0.7890 | −0.2662 |
| THR124, PHE125 | ASP313 | PHE226 | GLY316, ALA317 | ASP321, THR322 | LEU382 | ILE386 | THR498 | HEM900 | |
|---|---|---|---|---|---|---|---|---|---|
| ΔN | 0.035 | −0.846 | 0.0575 | 0.0554 | −0.7456 | 0.0545 | 0.0542 | 0.0431 | 0.6905 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Montoya, I.A.; Flores-Holguín, N.R.; Landeros-Martínez, L.-L.; Alvarado-González, M.; Rascón-Cruz, Q.; Fuentes-Montero, M.E.; Palomares-Báez, P.; Rodríguez-Valdez, L.M. Analysis of the Molecular Interactions between Cytochromes P450 3A4 and 1A2 and Aflatoxins: A Docking Study. Appl. Sci. 2019, 9, 2467. https://doi.org/10.3390/app9122467
García-Montoya IA, Flores-Holguín NR, Landeros-Martínez L-L, Alvarado-González M, Rascón-Cruz Q, Fuentes-Montero ME, Palomares-Báez P, Rodríguez-Valdez LM. Analysis of the Molecular Interactions between Cytochromes P450 3A4 and 1A2 and Aflatoxins: A Docking Study. Applied Sciences. 2019; 9(12):2467. https://doi.org/10.3390/app9122467
Chicago/Turabian StyleGarcía-Montoya, Isui Abril, Norma Rosario Flores-Holguín, Linda-Lucila Landeros-Martínez, Mónica Alvarado-González, Quintín Rascón-Cruz, María Elena Fuentes-Montero, Pedro Palomares-Báez, and Luz María Rodríguez-Valdez. 2019. "Analysis of the Molecular Interactions between Cytochromes P450 3A4 and 1A2 and Aflatoxins: A Docking Study" Applied Sciences 9, no. 12: 2467. https://doi.org/10.3390/app9122467
APA StyleGarcía-Montoya, I. A., Flores-Holguín, N. R., Landeros-Martínez, L.-L., Alvarado-González, M., Rascón-Cruz, Q., Fuentes-Montero, M. E., Palomares-Báez, P., & Rodríguez-Valdez, L. M. (2019). Analysis of the Molecular Interactions between Cytochromes P450 3A4 and 1A2 and Aflatoxins: A Docking Study. Applied Sciences, 9(12), 2467. https://doi.org/10.3390/app9122467