Directed Silica Co-Deposition by Highly Oxidized Silver: Enhanced Stability and Versatility of Silver Oxynitrate

Abstract
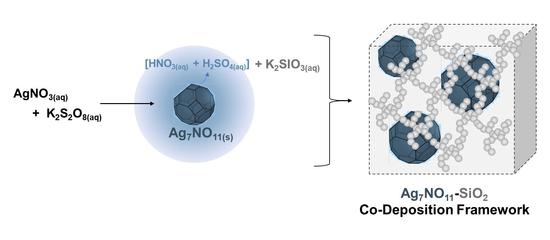
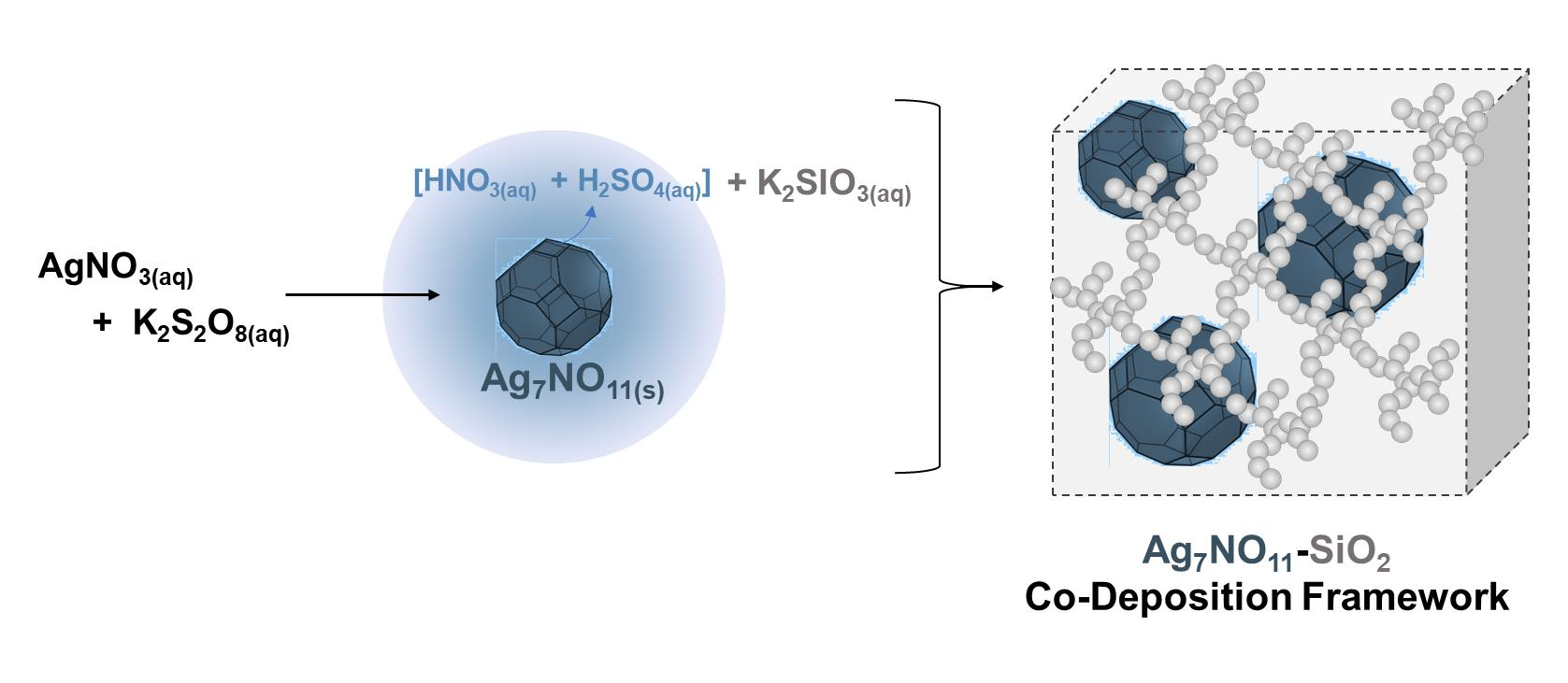
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Equipment
2.3. Preparation of the Ag7NO11:SiO2 Framework
2.4. Silver Quantification
2.5. X-Ray Diffractometry
2.6. Aqueous Decomposition Studies
2.7. Planktonic Log Reduction Assay
2.8. Biofilm Log Reduction Assay
2.9. Statistical Analysis
3. Results
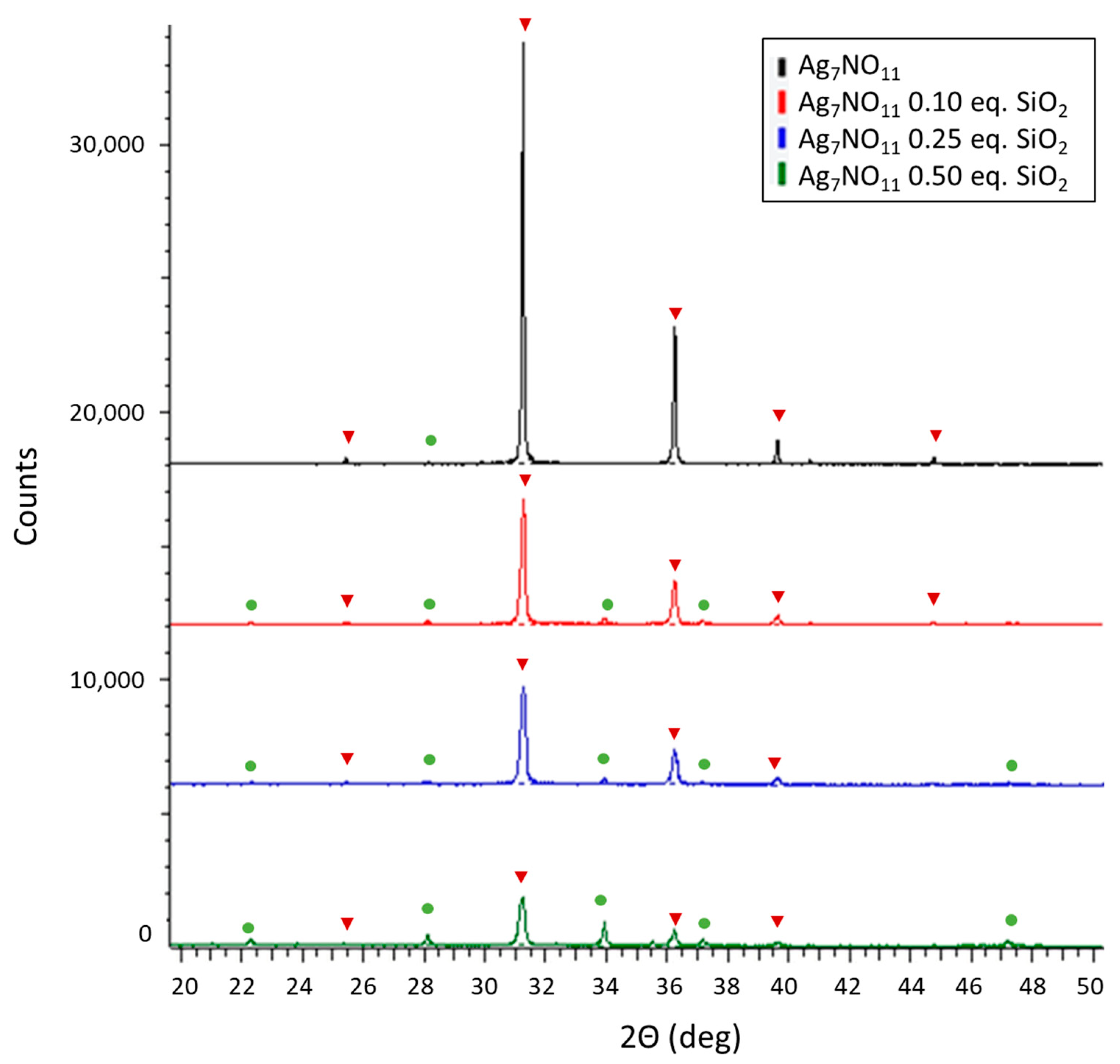
3.1. Characterization of Ag7NO11:SiO2
Ag7O8NO3 (s) + 6HNO3 (aq) + H2SO4 (aq) + K2SO4 (aq) + 4H2 (g)
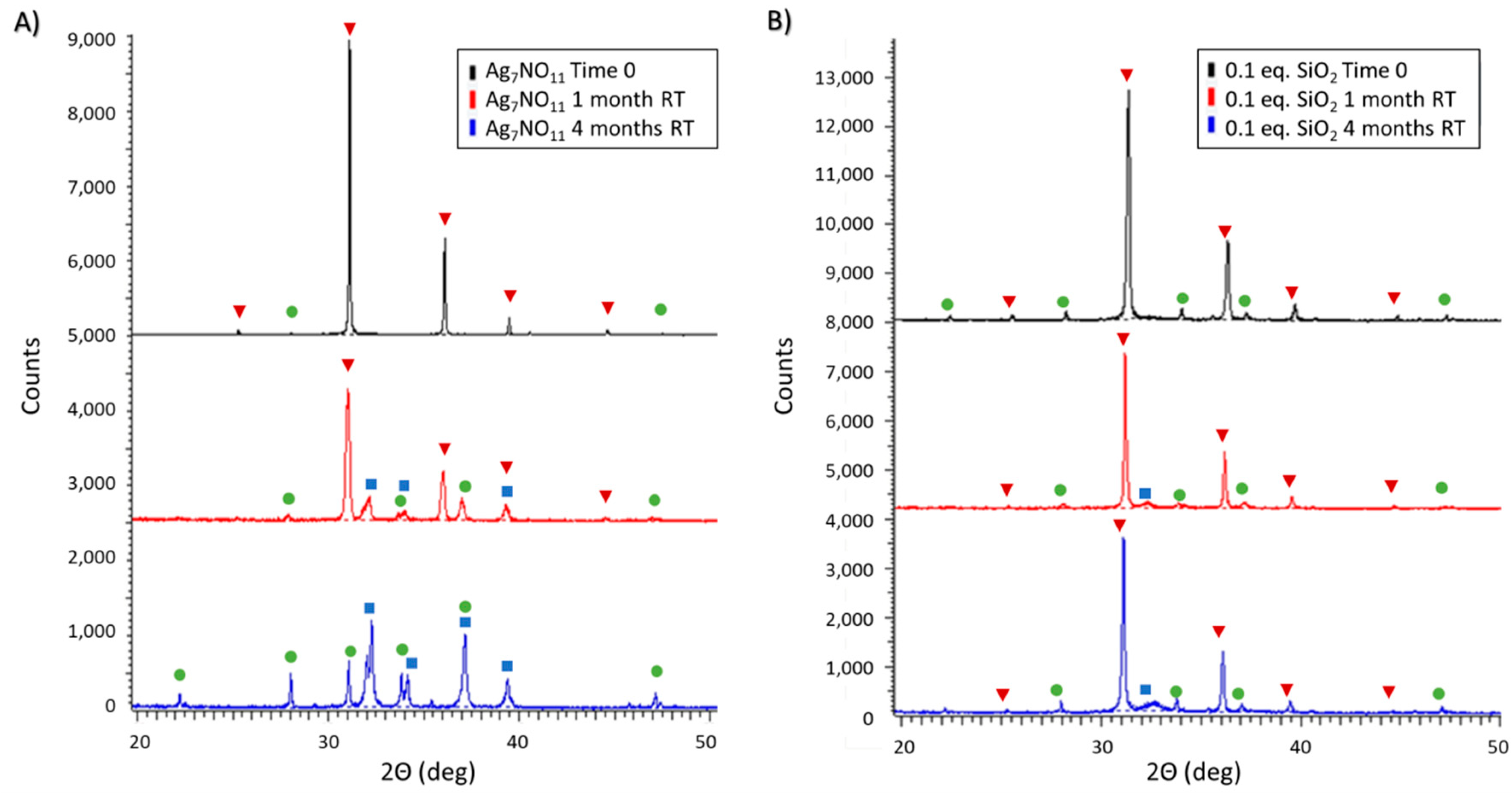
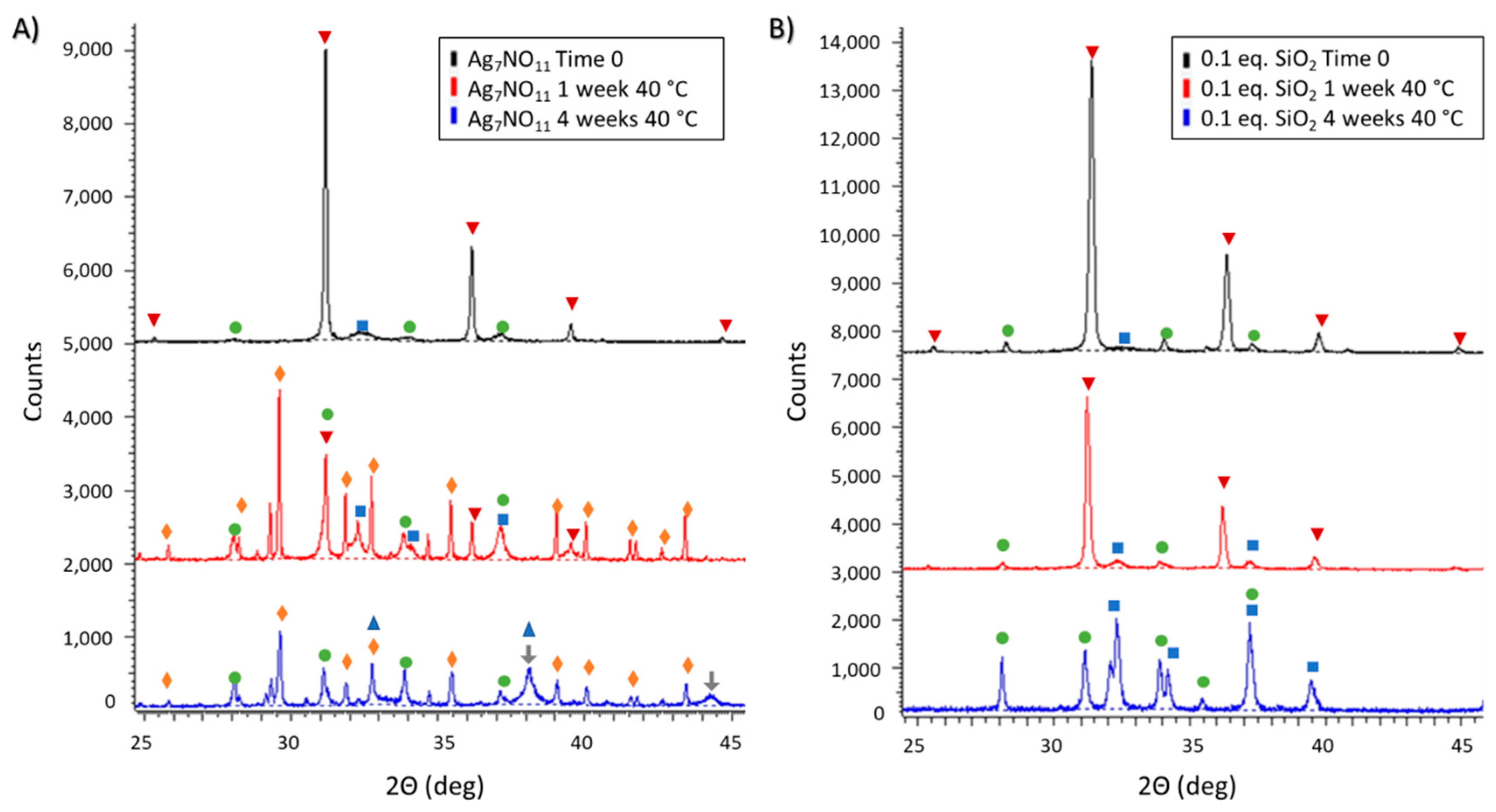
3.2. Ambient and Accelerated Thermal Stability of Ag7NO11:SiO2
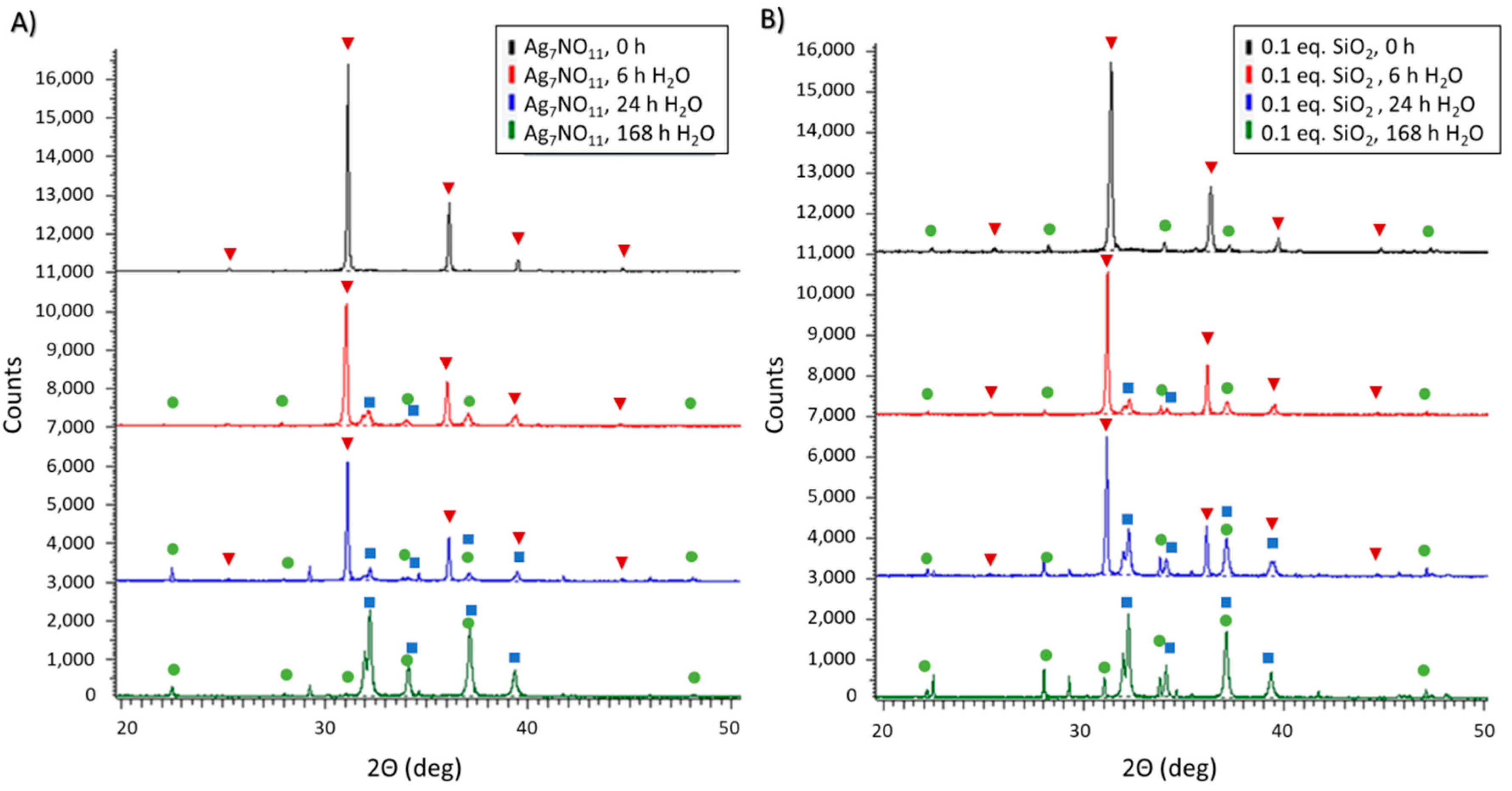
3.3. Aqueous Decomposition of Ag7NO11:SiO2
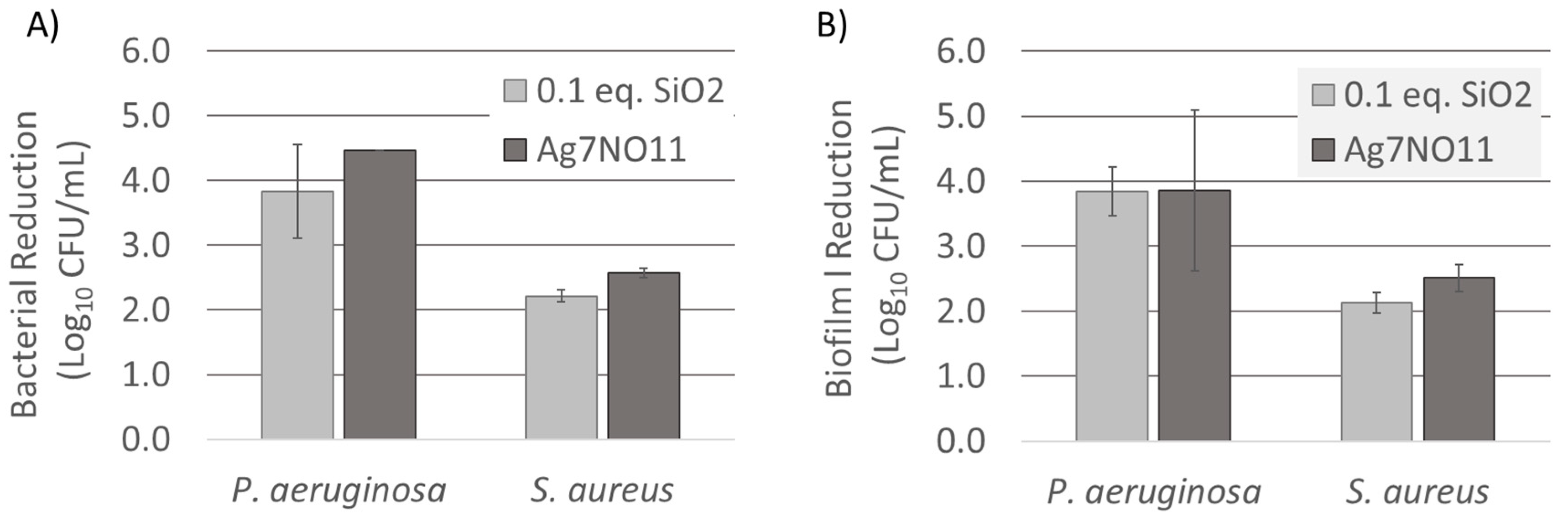
3.4. Antimicrobial Efficacy Evaluation of Ag7NO11:SiO2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Qian, K.K.; Bogner, R.H. Application of mesoporous silicon dioxide and silicate in oral amorphous drug delivery systems. J. Pharm. Sci. 2012, 101, 444–463. [Google Scholar] [CrossRef]
- BernArdos, A.; KouřimsKá, L. Applications of mesoporous silica materials in food-a review. Czech J. Food Sci. 2013, 31, 99–107. [Google Scholar] [CrossRef]
- Tang, H.; Xiong, H.; Tang, S.; Zou, P. A starch-based biodegradable film modified by nano silicon dioxide. J. Appl. Polym. Sci. 2009, 113, 34–40. [Google Scholar] [CrossRef]
- Santra, S.; Tapec, R.; Theodoropoulou, N.; Dobson, J.; Hebard, A.; Tan, W. Synthesis and characterization of silica-coated iron oxide nanoparticles in microemulsion: The effect of nonionic surfactants. Langmuir 2001, 17, 2900–2906. [Google Scholar] [CrossRef]
- Chung, C.C.; Jean, J.H. Aqueous Synthesis of Y2O2S: Eu/Silica Core-Shell Particles. J. Am. Ceram. Soc. 2005, 88, 1341–1344. [Google Scholar] [CrossRef]
- Stöber, W.; Fink, A.; Bohn, E. Controlled growth of monodisperse silica spheres in the micron size range. J. Colloid Interface Sci. 1968, 26, 62–69. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Katakami, H.; Mine, E.; Nagao, D.; Konno, M.; Liz-Marzán, L.M. Silica coating of silver nanoparticles using a modified Stöber method. J. Colloid Interface Sci. 2005, 283, 392–396. [Google Scholar] [CrossRef]
- Zhu, Y.; Shi, J.; Shen, W.; Dong, X.; Feng, J.; Ruan, M.; Li, Y. Stimuli-responsive controlled drug release from a hollow mesoporous silica sphere/polyelectrolyte multilayer core–shell structure. Angew. Chem. 2005, 117, 5213–5217. [Google Scholar] [CrossRef]
- Xu, K.; Wang, J.X.; Kang, X.L.; Chen, J.F. Fabrication of antibacterial monodispersed Ag–SiO2 core–shell nanoparticles with high concentration. Mater. Lett. 2009, 63, 31–33. [Google Scholar] [CrossRef]
- Klasen, H.J. A historical review of the use of silver in the treatment of burns. II. Renewed interest for silver. Burns 2000, 26, 131–138. [Google Scholar] [CrossRef]
- Barillo, D.J.; Marx, D.E. Silver in medicine: A brief history BC 335 to present. Burns 2014, 40, S3–S8. [Google Scholar] [CrossRef]
- Alexander, J.W. History of the medical use of silver. Surg. Infect. 2009, 10, 289–292. [Google Scholar] [CrossRef]
- Lansdown, A.B. Silver I: Its antibacterial properties and mechanism of action. J. Wound Care 2002, 11, 125–130. [Google Scholar] [CrossRef]
- Jung, W.K.; Koo, H.C.; Kim, K.W.; Shin, S.; Kim, S.H.; Park, Y.H. Antibacterial activity and mechanism of action of the silver ion in Staphylococcus aureus and Escherichia coli. Appl. Environ. Microbiol. 2008, 74, 2171–2178. [Google Scholar] [CrossRef]
- Holt, K.B.; Bard, A.J. Interaction of silver (I) ions with the respiratory chain of Escherichia coli: An electrochemical and scanning electrochemical microscopy study of the antimicrobial mechanism of micromolar Ag+. Biochemistry 2005, 44, 13214–13223. [Google Scholar] [CrossRef]
- Lemire, J.A.; Kalan, L.; Bradu, A.; Turner, R.J. Silver oxynitrate, an unexplored silver compound with antimicrobial and antibiofilm activity. Antimicrob. Agents Chemother. 2015, 59, 4031–4039. [Google Scholar] [CrossRef]
- Lemire, J.A.; Kalan, L.; Gugala, N.; Bradu, A.; Turner, R.J. Silver oxynitrate–an efficacious compound for the prevention and eradication of dual-species biofilms. Biofouling 2017, 33, 460–469. [Google Scholar] [CrossRef]
- Kalan, L.R.; Pepin, D.M.; Ul-Haq, I.; Miller, S.B.; Hay, M.E.; Precht, R.J. Targeting biofilms of multidrug-resistant bacteria with silver oxynitrate. Int. J. Antimicrob. Agents 2017, 49, 719–726. [Google Scholar] [CrossRef]
- Thomason, H.A.; Lovett, J.M.; Spina, C.J.; Stephenson, C.; McBain, A.J.; Hardman, M.J. Silver oxysalts promote cutaneous wound healing independent of infection. Wound Repair Regen. 2018, 26, 144–152. [Google Scholar] [CrossRef]
- Vanysek, P. Electrochemical series. In CRC Handbook of Chemistry and Physics; CRC Press: Boca Raton, FL, USA, 2000; p. 8. [Google Scholar]
- Jack, J.C.; Kennedy, T. The thermal analysis of “argentic oxynitrate” and silver oxides. J. Anal. 1971, 3, 25–33. [Google Scholar] [CrossRef]
- Waterhouse, G.I.; Bowmaker, G.A.; Metson, J.B. The thermal decomposition of silver (I, III) oxide: A combined XRD, FT-IR and Raman spectroscopic study. Phys. Chem. Chem. Phys. 2001, 3, 3838–3845. [Google Scholar] [CrossRef]
- Waterhouse, G.I.; Metson, J.B.; Bowmaker, G.A. Synthesis, vibrational spectra and thermal stability of Ag3O4 and related Ag7O8X salts (X = NO3-, ClO4-, HSO4-). Polyhedron 2007, 26, 3310–3322. [Google Scholar] [CrossRef]
- Bałdyga, J.; Jasińska, M.; Jodko, K.; Petelski, P. Precipitation of amorphous colloidal silica from aqueous solutions—Aggregation problem. Chem. Eng. Sci. 2012, 77, 207–216. [Google Scholar] [CrossRef]
- Musić, S.; Filipović-Vinceković, N.; Sekovanić, L. Precipitation of amorphous SiO2 particles and their properties. Braz. J. Chem. Eng. 2011, 28, 89–94. [Google Scholar] [CrossRef]
- Belton, D.J.; Deschaume, O.; Perry, C.C. An overview of the fundamentals of the chemistry of silica with relevance to biosilicification and technological advances. FEBS J. 2012, 279, 1710–1720. [Google Scholar] [CrossRef]
- Ray, R.C.; Ganguly, P.B. The optimum conditions for the formation of silica gels from alkali silicate solutions. J. Phys. Chem. 2002, 34, 352–358. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Sakuraba, T. Silica-coating of metallic copper nanoparticles in aqueous solution. Colloids Surf. A Physicochem. Eng. Asp. 2008, 317, 756–759. [Google Scholar] [CrossRef]
- Naray-Szabo, I.; Argay, G.; Szabo, P. The structure of the Ag (I, III) oxide phases. Acta Crystallogr. 1965, 19, 180–184. [Google Scholar] [CrossRef]
- Djokić, S.S. Deposition of silver oxysalts and their antimicrobial properties. J. Electrochem. Soc. 2004, 151, C359–C364. [Google Scholar] [CrossRef]
- Hou, X.; Kirkpatrick, R.J.; Struble, L.J.; Monteiro, P.J. Structural investigations of alkali silicate gels. J. Am. Ceram. Soc. 2005, 88, 943–949. [Google Scholar] [CrossRef]
- Klug, H.P.; Alexander, L.E. X-ray diffraction procedures: For polycrystalline and amorphous materials. In X-Ray Diffraction Procedures: For Polycrystalline and Amorphous Materials, 2nd ed.; Klug, H.P., Alexander, L.E., Eds.; Wiley-VCH: Hoboken, NJ, USA, 1974; p. 992. ISBN 0-471-49369-4. [Google Scholar]
- Schierle, C.F.; De la Garza, M.; Mustoe, T.A.; Galiano, R.D. Staphylococcal biofilms impair wound healing by delaying reepithelialization in a murine cutaneous wound model. Wound Repair Regen. 2009, 17, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, D.G.; Bowler, P.G. Biofilm delays wound healing: A review of the evidence. Burn. Trauma 2013, 1, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolcott, R.D.; Rhoads, D.D.; Dowd, S.E. Biofilms and chronic wound inflammation. J. Wound Care 2008, 17, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Seth, A.K.; Zhong, A.; Nguyen, K.T.; Hong, S.J.; Leung, K.P.; Galiano, R.D.; Mustoe, T.A. Impact of a novel, antimicrobial dressing on in vivo, Pseudomonas aeruginosa wound biofilm: Quantitative comparative analysis using a rabbit ear model. Wound Repair Regen 2014, 22, 712–719. [Google Scholar] [CrossRef]
- Cong, L.; Takeda, M.; Hamanaka, Y.; Gonda, K.; Watanabe, M.; Kumasaka, M.; Kobayashi, Y.; Kobayashi, M.; Ohuchi, N. Uniform silica coated fluorescent nanoparticles: Synthetic method, improved light stability and application to visualize lymph network tracer. PLoS ONE 2010, 5, e13167. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.; Li, L.; Chen, D. Mesoporous silica nanoparticles: Synthesis, biocompatibility and drug delivery. Adv. Mater. 2012, 24, 1504–1534. [Google Scholar] [CrossRef]
- Barbe, C.; Bartlett, J.; Kong, L.; Finnie, K.; Lin, H.Q.; Larkin, M.; Calleja, S.; Bush, A.; Calleja, G. Silica particles: A novel drug-delivery system. Adv. Mater. 2004, 16, 1959–1966. [Google Scholar] [CrossRef]
- Makrides, A.C.; Turner, M.; Slaughter, J. Condensation of silica from supersaturated silicic acid solutions. J. Colloid Interface Sci. 1980, 73, 345–367. [Google Scholar] [CrossRef]
- McMillan, J.A. Higher Oxidation States of Silver. Chem. Rev. 1962, 62, 65–80. [Google Scholar] [CrossRef]
- Holzwarth, U.; Gibson, N. The Scherrer equation versus the’Debye-Scherrer equation’. Nat. Nanotechnol. 2011, 6, 534. [Google Scholar] [CrossRef]
- Schmidt, M.; Schwertfeger, F. Applications for silica aerogel products. J. Non-Cryst. Solids 1998, 225, 364–368. [Google Scholar] [CrossRef]
- Reim, M.; Körner, W.; Manara, J.; Korder, S.; Arduini-Schuster, M.; Ebert, H.P.; Fricke, J. Silica aerogel granulate material for thermal insulation and daylighting. Sol. Energy 2005, 79, 131–139. [Google Scholar] [CrossRef]
- Sandberg, L.I.; Gao, T.; Jelle, B.P.; Gustavsen, A. Synthesis of hollow silica nanospheres by sacrificial polystyrene templates for thermal insulation applications. Adv. Mater. Sci. Eng. 2013, 2013, 483651. [Google Scholar] [CrossRef] [Green Version]
- Williams, D.B.; Lawton, M. Drying of organic solvents: Quantitative evaluation of the efficiency of several desiccants. J. Org. Chem. 2010, 75, 8351–8354. [Google Scholar] [CrossRef]
- Sherlock, G.; Block, W.; Benson, E.E. Thermal analysis of the plant encapsulation-dehydration cryopreservation protocol using silica gel as the desiccant. CryoLetters 2005, 26, 45–54. [Google Scholar]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spina, C.J.; Ladhani, R.; Goodall, C.; Hay, M.; Precht, R. Directed Silica Co-Deposition by Highly Oxidized Silver: Enhanced Stability and Versatility of Silver Oxynitrate. Appl. Sci. 2019, 9, 5236. https://doi.org/10.3390/app9235236
Spina CJ, Ladhani R, Goodall C, Hay M, Precht R. Directed Silica Co-Deposition by Highly Oxidized Silver: Enhanced Stability and Versatility of Silver Oxynitrate. Applied Sciences. 2019; 9(23):5236. https://doi.org/10.3390/app9235236
Chicago/Turabian StyleSpina, Carla J., Roohee Ladhani, Carlie Goodall, Michelle Hay, and Rod Precht. 2019. "Directed Silica Co-Deposition by Highly Oxidized Silver: Enhanced Stability and Versatility of Silver Oxynitrate" Applied Sciences 9, no. 23: 5236. https://doi.org/10.3390/app9235236
APA StyleSpina, C. J., Ladhani, R., Goodall, C., Hay, M., & Precht, R. (2019). Directed Silica Co-Deposition by Highly Oxidized Silver: Enhanced Stability and Versatility of Silver Oxynitrate. Applied Sciences, 9(23), 5236. https://doi.org/10.3390/app9235236