A Theoretical Study of the Energetic Stability and Geometry of Silicon-Vacancy Color Centers in Diamond (001) Surfaces

Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
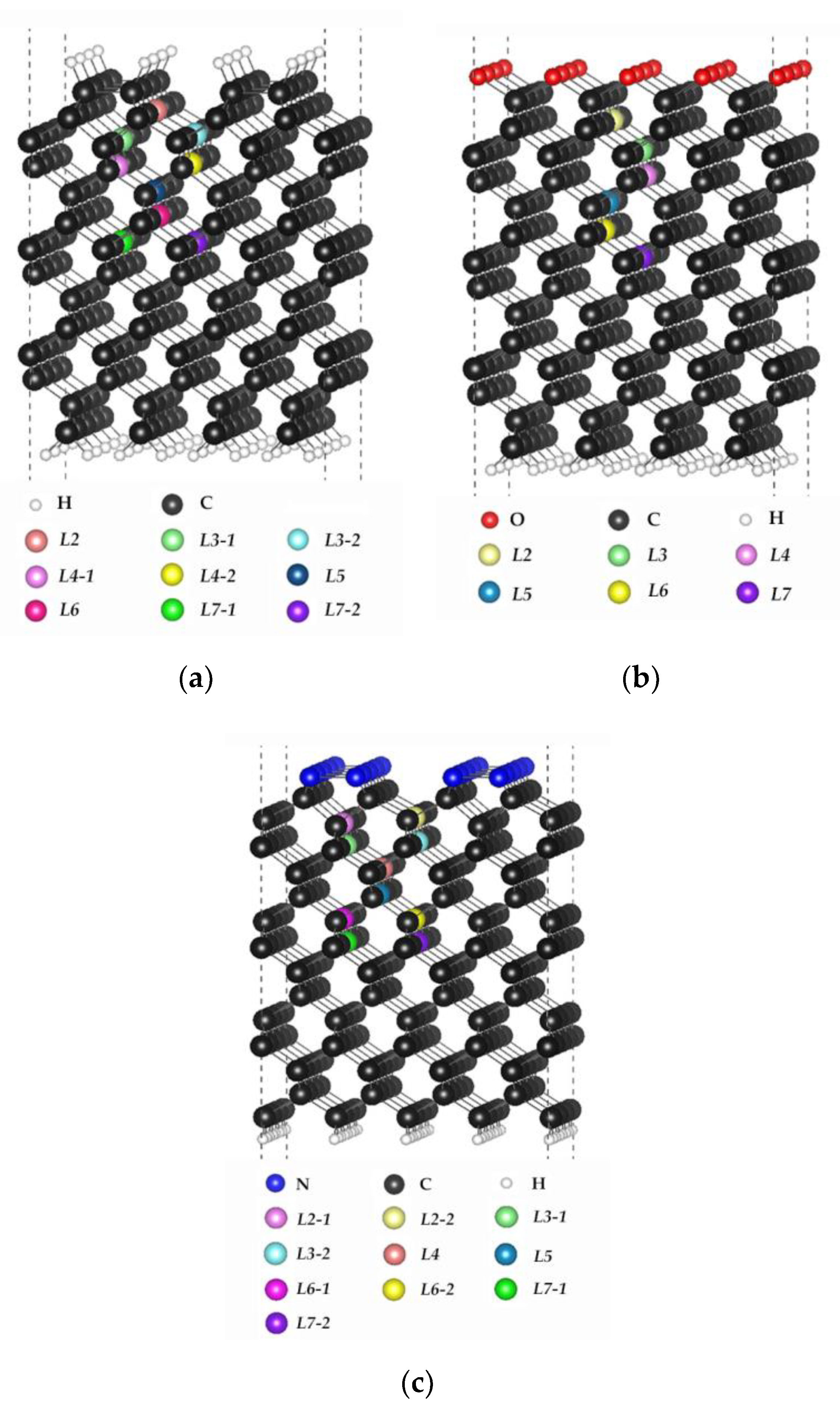
3.1. Single Substitutional Silicon Impurity in H-, O-, and N-Terminated C (001) Surfaces
3.2. Single Vacancy in H-, O-, or N-Terminated C (001) Surfaces
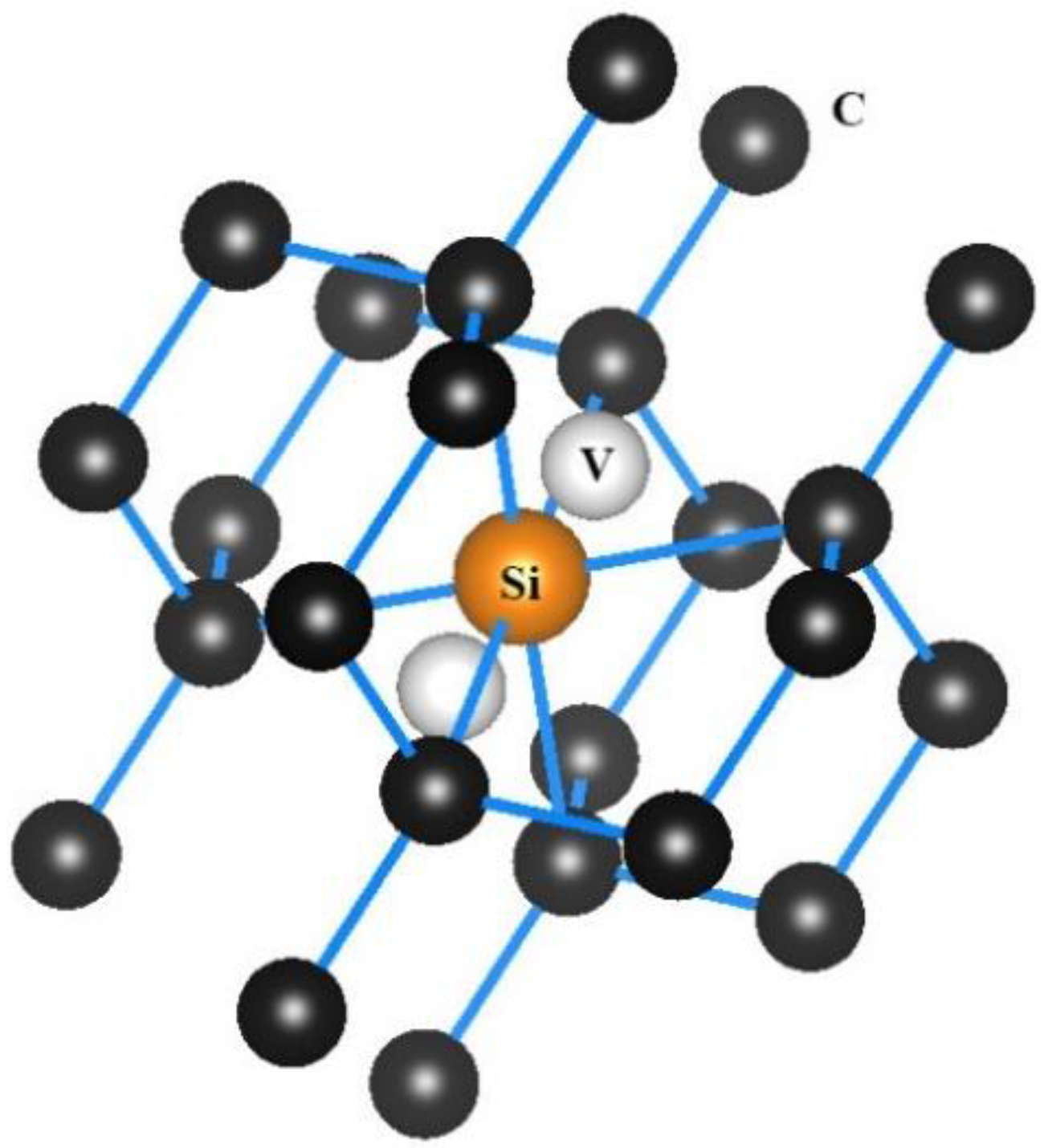
3.3. Single Neutral Silicon Vacancy in the H-, O-, or N-Terminated C (001) Surfaces
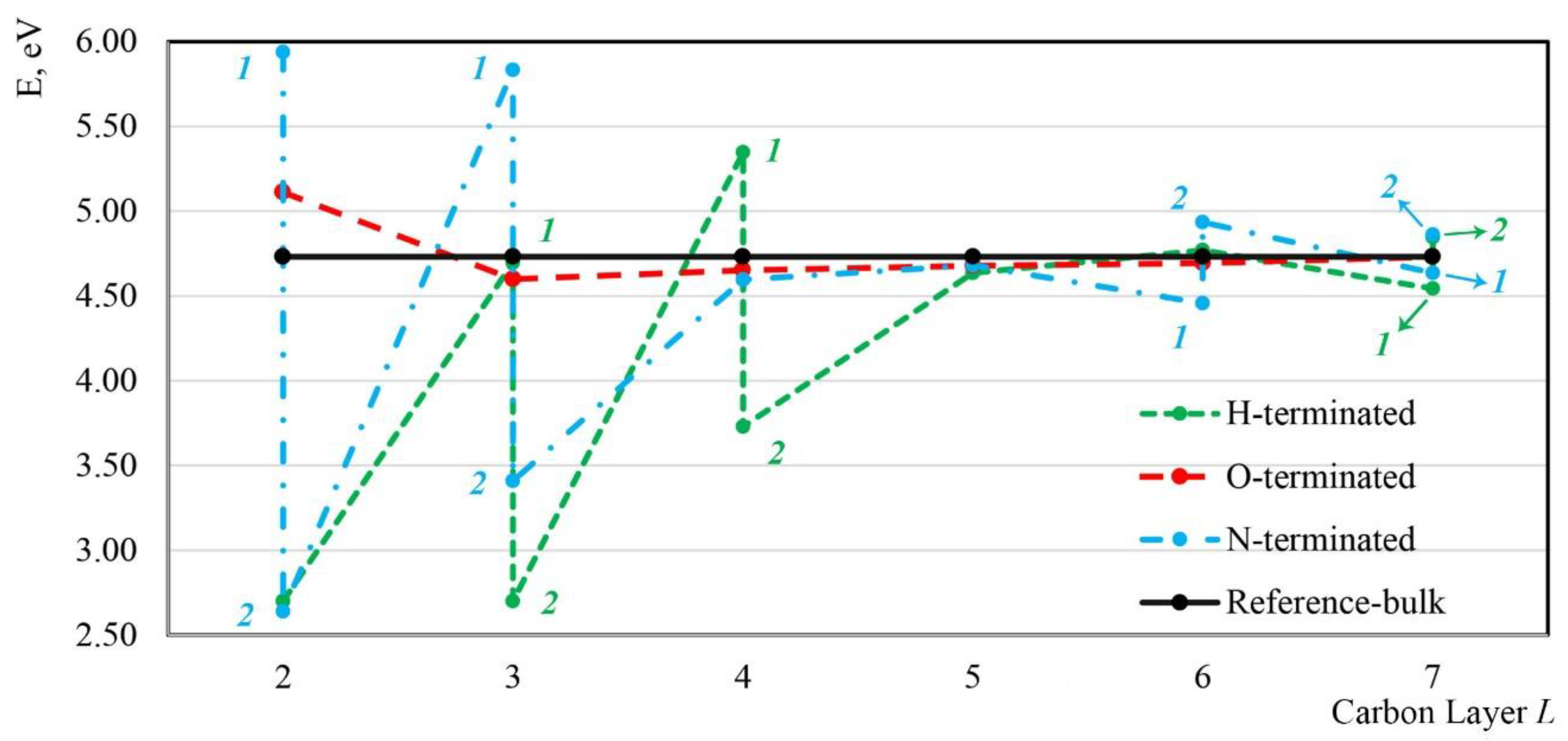
3.3.1. Formation Energies
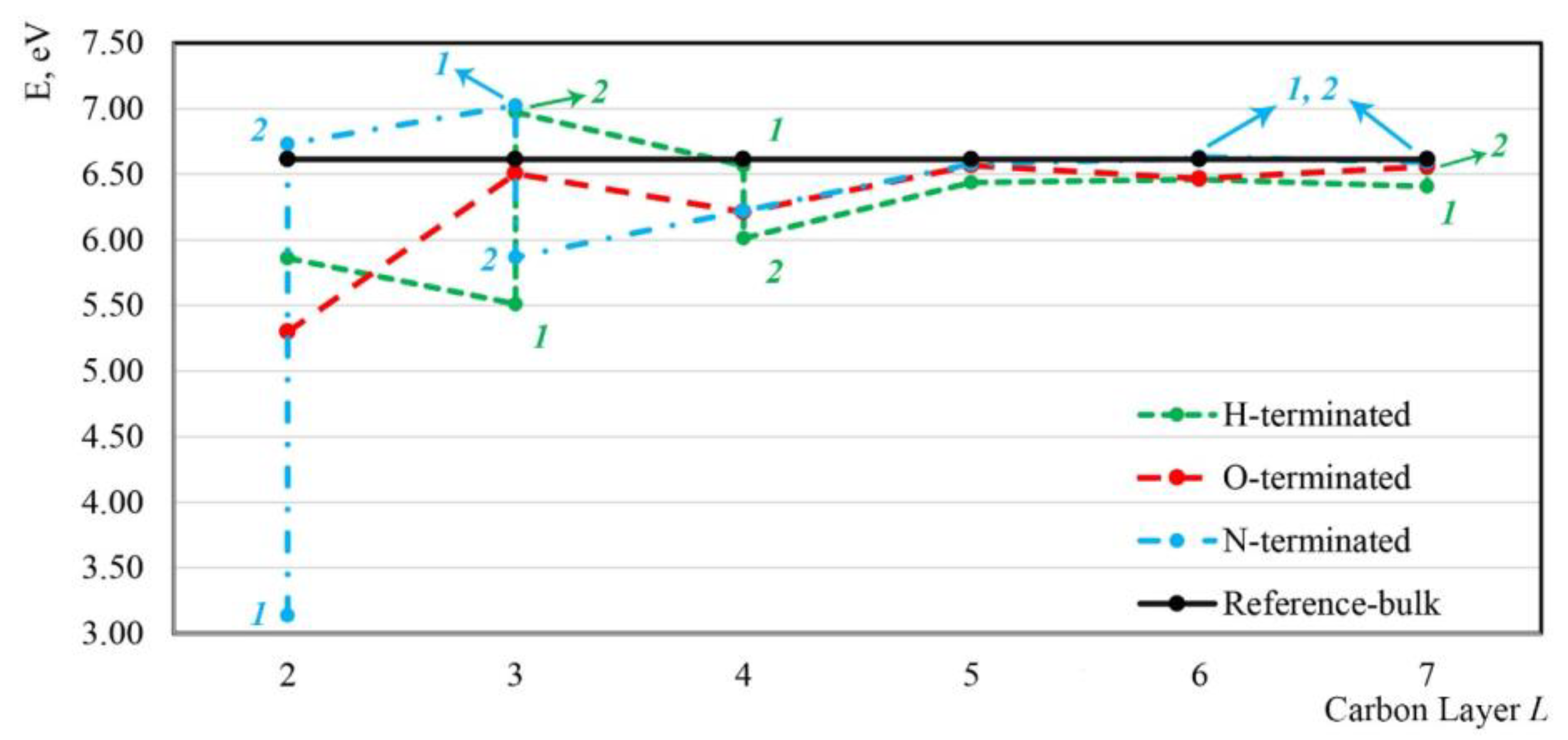
3.3.2. Binding Energies
3.3.3. Geometric Structures
3.3.4. Density of States
4. Conclusions
- (1).
- The effects of the termination species on inner diamond atoms decayed to be negligible below the fifth C layer. The formation energies of under H-, O-, or N-terminated diamond (001) surfaces converged to the corresponding value in bulk diamond when they were positioned below the fifth C layer. Additionally, when deeper than the fifth C layer, regardless of H, O, or N termination, the C–C bond lengths reached a stable value, almost the same as that in bulk diamond.
- (2).
- Regarding formation energy, it was generally easier for to be embedded in an O-terminated diamond (001) surface when this was above the fifth C layer. Formation energies were almost the same when placed below the fifth C layer among H, O, and N terminations. However, it was similarly easy for to form as both configuration 1 (i.e., its original (prior to geometry optimization) Si or V site was positioned in the third C layer, below the dimer rows) and configuration 2 (i.e., its original (prior to geometry optimization) Si or V site was positioned in the third C layer, between the dimer rows) at the second and to third layers under N termination, and as configuration 2 (i.e., both of its original (prior to geometry optimization) Si and V site were positioned in the third or fourth C layers, between the dimer rows) at the third and fourth layers under H termination as compared with O termination.
- (3).
- Si generally showed a lower formation energy than SiV near H-, O-, or N-terminated diamond (001) surfaces. However, SiV centers exhibited rather high energetic stability once formed, as indicated by their binding energies.
- (4).
- H termination exhibited a contracting effect on inner surface atoms. For O termination, there was a small expanding effect on inner surface atoms. Similar to H termination, N termination showed a small contracting effect on inner surface atoms.
- (5).
- H termination species introduced heavy impurity energy levels deep down in the CBM, and even worse, deep impurity energy levels were introduced not only above the VBM but also below CBM under O termination, in the DOS of diamond (001) surfaces. Our calculations revealed that there were almost no such impurity energy levels above the VBM or below the CBM for N termination, which indicates that an N-terminated diamond (001) surface is a suitable medium for the use of single as a single-photon source.
Author Contributions
Funding
Conflicts of Interest
References
- Schirhagl, R.; Chang, K.; Loretz, M.; Degen, C.L. Nitrogen-vacancy centers in diamond: Nanoscale sensors for physics and biology. Annu. Rev. Phys. Chem. 2014, 65, 83–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, W.; Pan, Y.; Shen, S.; Li, H.; Zhang, Y.; Zhang, G. Electron affinity of boron-terminated diamond (001) surfaces: A density functional theory study. J. Mater. Chem. C 2019, 7, 9756–9765. [Google Scholar] [CrossRef]
- Knill, E.; Laflamme, R.; Milburn, G.J. A scheme for efficient quantum computation with linear optics. Nature 2001, 409, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Gisin, N.; Ribordy, G.; Tittel, W.; Zbinden, H. Quantum cryptography. Rev. Mod. Phys. 2002, 74, 145–195. [Google Scholar] [CrossRef] [Green Version]
- Neumann, P.; Kolesov, R.; Naydenov, B.; Beck, J.; Rempp, F.; Steiner, M.; Jacques, V.; Balasubramanian, G.; Markham, M.L.; Twitchen, D.J.; et al. Quantum register based on coupled electron spins in a room-temperature solid. Nat. Phys. 2010, 6, 249. [Google Scholar] [CrossRef]
- Maletinsky, P.; Hong, S.; Grinolds, M.S.; Hausmann, B.; Lukin, M.D.; Walsworth, R.L.; Loncar, M.; Yacoby, A. A robust scanning diamond sensor for nanoscale imaging with single nitrogen-vacancy centres. Nat. Nanotechnol. 2012, 7, 320. [Google Scholar] [CrossRef]
- Rondin, L.; Tetienne, J.P.; Hingant, T.; Roch, J.F.; Maletinsky, P.; Jacques, V. Magnetometry with nitrogen-vacancy defects in diamond. Rep. Prog. Phys. 2014, 77, 056503. [Google Scholar] [CrossRef] [Green Version]
- McGuinness, L.P.; Yan, Y.; Stacey, A.; Simpson, D.A.; Hall, L.T.; Maclaurin, D.; Prawer, S.; Mulvaney, P.; Wrachtrup, J.; Caruso, F.; et al. Quantum measurement and orientation tracking of fluorescent nanodiamonds inside living cells. Nat. Nanotechnol. 2011, 6, 358. [Google Scholar] [CrossRef]
- Le Sage, D.; Arai, K.; Glenn, D.R.; DeVience, S.J.; Pham, L.M.; Rahn-Lee, L.; Lukin, M.D.; Yacoby, A.; Komeili, A.; Walsworth, R.L. Optical magnetic imaging of living cells. Nature 2013, 496, 486. [Google Scholar] [CrossRef]
- Aharonovich, I.; Castelletto, S.; Simpson, D.A.; Su, C.H.; Greentree, A.D.; Prawer, S. Diamond-based single-photon emitters. Rep. Prog. Phys. 2011, 74, 076501. [Google Scholar] [CrossRef]
- Pezzagna, S.; Rogalla, D.; Wildanger, D.; Meijer, J.; Zaitsev, A. Creation and nature of optical centres in diamond for single-photon emission—Overview and critical remarks. New J. Phys. 2011, 13, 035024. [Google Scholar] [CrossRef]
- Brouri, R.; Beveratos, A.; Poizat, J.-P.; Grangier, P. Photon antibunching in the fluorescence of individual color centers in diamond. Opt. Lett. 2000, 25, 1294–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuochi, N.; Makino, T.; Kato, H.; Takeuchi, D.; Ogura, M.; Okushi, H.; Nothaft, M.; Neumann, P.; Gali, A.; Jelezko, F.; et al. Electrically driven single-photon source at room temperature in diamond. Nat. Photonics 2012, 6, 299. [Google Scholar] [CrossRef]
- Wang, C.; Kurtsiefer, C.; Weinfurter, H.; Burchard, B. Single photon emission from SiV centres in diamond produced by ion implantation. J. Phys. B 2005, 39, 37–41. [Google Scholar] [CrossRef] [Green Version]
- Neu, E.; Steinmetz, D.; Riedrich-Möller, J.; Gsell, S.; Fischer, M.; Schreck, M.; Becher, C. Single photon emission from silicon-vacancy colour centres in chemical vapour deposition nano-diamonds on iridium. New J. Phys. 2011, 13, 025012. [Google Scholar] [CrossRef]
- Gaebel, T.; Popa, I.; Gruber, A.; Domhan, M.; Jelezko, F.; Wrachtrup, J. Stable single-photon source in the near infrared. New J. Phys. 2004, 6, 98. [Google Scholar] [CrossRef]
- Rabeau, J.R.; Chin, Y.L.; Prawer, S.; Jelezko, F.; Gaebel, T.; Wrachtrup, J. Fabrication of single nickel-nitrogen defects in diamond by chemical vapor deposition. Appl. Phys. Lett. 2005, 86, 131926. [Google Scholar] [CrossRef] [Green Version]
- Aharonovich, I.; Castelletto, S.; Simpson, D.A.; Stacey, A.; McCallum, J.; Greentree, A.D.; Prawer, S. Two-level ultrabright single photon emission from diamond nanocrystals. Nano Lett. 2009, 9, 3191–3195. [Google Scholar] [CrossRef]
- Iwasaki, T.; Ishibashi, F.; Miyamoto, Y.; Doi, Y.; Kobayashi, S.; Miyazaki, T.; Tahara, K.; Jahnke, K.D.; Rogers, L.J.; Naydenov, B.; et al. Germanium-vacancy single color centers in diamond. Sci. Rep. 2015, 5, 12882. [Google Scholar] [CrossRef] [Green Version]
- Neumann, P.; Kolesov, R.; Jacques, V.; Beck, J.; Tisler, J.; Batalov, A.; Rogers, L.; Manson, N.B.; Balasubramanian, G.; Jelezko, F.; et al. Excited-state spectroscopy of single nv defects in diamond using optically detected magnetic resonance. New J. Phys. 2009, 11, 013017. [Google Scholar] [CrossRef]
- Tisler, J.; Balasubramanian, G.; Naydenov, B.; Kolesov, R.; Grotz, B.; Reuter, R.; Boudou, J.-P.; Curmi, P.A.; Sennour, M.; Thorel, A.; et al. Fluorescence and spin properties of defects in single digit nanodiamonds. ACS Nano 2009, 3, 1959–1965. [Google Scholar] [CrossRef] [PubMed]
- Lim, T.-S.; Fu, C.-C.; Lee, K.-C.; Lee, H.-Y.; Chen, K.; Cheng, W.-F.; Pai, W.W.; Chang, H.-C.; Fann, W. Fluorescence enhancement and lifetime modification of single nanodiamonds near a nanocrystalline silver surface. PCCP 2009, 11, 1508–1514. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, A.; Jahnke, K.D.; Binder, J.M.; Teraji, T.; Isoya, J.; Rogers, L.J.; Jelezko, F. Isotopically varying spectral features of silicon-vacancy in diamond. New J. Phys. 2014, 16, 113019. [Google Scholar] [CrossRef]
- Sternschulte, H.; Thonke, K.; Sauer, R.; Münzinger, P.C.; Michler, P. 1.681-ev luminescence center in chemical-vapor-deposited homoepitaxial diamond films. Phys. Rev. B 1994, 50, 14554–14560. [Google Scholar] [CrossRef]
- Rose, B.C.; Huang, D.; Zhang, Z.-H.; Stevenson, P.; Tyryshkin, A.M.; Sangtawesin, S.; Srinivasan, S.; Loudin, L.; Markham, M.L.; Edmonds, A.M.; et al. Observation of an environmentally insensitive solid-state spin defect in diamond. Science 2018, 361, 60. [Google Scholar] [CrossRef] [Green Version]
- Vlasov, I.I.; Barnard, A.S.; Ralchenko, V.G.; Lebedev, O.I.; Kanzyuba, M.V.; Saveliev, A.V.; Konov, V.I.; Goovaerts, E. Nanodiamond photoemitters based on strong narrow-band luminescence from silicon-vacancy defects. Adv. Mater. 2009, 21, 808–812. [Google Scholar] [CrossRef]
- Shershulin, V.A.; Sedov, V.S.; Ermakova, A.; Jantzen, U.; Rogers, L.; Huhlina, A.A.; Teverovskaya, E.G.; Ralchenko, V.G.; Jelezko, F.; Vlasov, I.I. Size-dependent luminescence of color centers in composite nanodiamonds. Phys. Status Solidi A 2015, 212, 2600–2605. [Google Scholar] [CrossRef]
- Bolshakov, A.; Ralchenko, V.; Sedov, V.; Khomich, A.; Vlasov, I.; Khomich, A.; Trofimov, N.; Krivobok, V.; Nikolaev, S.; Khmelnitskii, R.; et al. Photoluminescence of SiV centers in single crystal CVD diamond in situ doped with Si from silane. Phys. Status Solidi A 2015, 212, 2525–2532. [Google Scholar] [CrossRef]
- Stehlik, S.; Ondic, L.; Varga, M.; Fait, J.; Artemenko, A.; Glatzel, T.; Kromka, A.; Rezek, B. Silicon-vacancy centers in ultra-thin nanocrystalline diamond films. Micromachines 2018, 9, 281. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Li, J.; Guo, L.; Huang, N.; Liu, L.; Zhai, Z.; Long, W.; Jiang, X. Fabrication of silicon-vacancy color centers in diamond films: Tetramethylsilane as a new dopant source. CrystEngComm 2018, 20, 1158–1167. [Google Scholar] [CrossRef]
- Kim, H.; Kim, H.; Lee, J.; Lim, W.C.; Eliades, J.A.; Kim, J.; Song, J.; Suk, J. Fabrication of silicon-vacancy color centers in nanodiamonds by using si-ion implantation. J. Korean Phys. Soc. 2018, 73, 661–666. [Google Scholar] [CrossRef]
- Rong, Y.; Cheng, K.; Ju, Z.; Pan, C.; Ma, Q.; Liu, S.; Shen, S.; Wu, B.; Jia, T. Bright near-surface silicon vacancy centers in diamond fabricated by femtosecond laser ablation. Opt. Lett. 2019, 44, 3793–3796. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Fan, D.; Lu, S.; Shen, Y.; Hu, X. Siv center photoluminescence induced by C=O termination in nanocrystalline diamond and graphite loops hybridized films. J. Appl. Phys. 2016, 120, 225107. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.; Refson, K.; Payne, M.C. First principles methods using castep. Zeitschrift für Kristallographie 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Riley, D.P. Lattice constant of diamond and the C–C single bond. Nature 1944, 153, 587–588. [Google Scholar] [CrossRef]
- Pfrommer, B.G.; Côté, M.; Louie, S.G.; Cohen, M.L. Relaxation of crystals with the quasi-newton method. J. Comput. Phys. 1997, 131, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Van de Walle, C.G.; Neugebauer, J. First-principles calculations for defects and impurities: Applications to III-nitrides. J. Appl. Phys. 2004, 95, 3851–3879. [Google Scholar] [CrossRef]
- Halicioglu, T. Calculations for defects formed on diamond surfaces. Thin Solid Films 1993, 228, 293–296. [Google Scholar] [CrossRef]
- O’Donnell, K.M.; Martin, T.L.; Fox, N.A.; Cherns, D. Ab initio investigation of lithium on the diamond C(100) surface. Phys. Rev. B 2010, 82, 115303. [Google Scholar] [CrossRef] [Green Version]
- Petrini, D.; Larsson, K. A theoretical study of the energetic stability and geometry of hydrogen- and oxygen-terminated diamond (100) surfaces. J. Phys. Chem. C 2007, 111, 795–801. [Google Scholar] [CrossRef]
- Shen, W.; Shen, S.; Liu, S.; Li, H.; Nie, S.; Pan, Y.; Tian, Z.; Li, Q. Binding of hydrogen to phosphorus dopant in phosphorus-doped diamond surfaces: A density functional theory study. Appl. Surf. Sci. 2019, 471, 309–317. [Google Scholar] [CrossRef]
- Bartell, L.S. On the length of the carbon-carbon single bond1. J. Am. Chem. Soc. 1959, 81, 3497–3498. [Google Scholar] [CrossRef]
- Zywietz, A.; Furthmüller, J.; Bechstedt, F. Neutral vacancies in group-IV semiconductors. Phys. Status Solidi B 1998, 210, 13–29. [Google Scholar] [CrossRef]
- Hood, R.Q.; Kent, P.R.C.; Needs, R.J.; Briddon, P.R. Quantum Monte Carlo study of the optical and diffusive properties of the vacancy defect in diamond. Phys. Rev. Lett. 2003, 91, 076403. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Salter, P.S.; Knauer, S.; Weng, L.; Frangeskou, A.C.; Stephen, C.J.; Ishmael, S.N.; Dolan, P.R.; Johnson, S.; Green, B.L.; et al. Laser writing of coherent colour centres in diamond. Nat. Photonics 2016, 11, 77. [Google Scholar] [CrossRef] [Green Version]
- Kononenko, V.V.; Vlasov, I.I.; Gololobov, V.M.; Kononenko, T.V.; Semenov, T.A.; Khomich, A.A.; Shershulin, V.A.; Krivobok, V.S.; Konov, V.I. Nitrogen-vacancy defects in diamond produced by femtosecond laser nanoablation technique. Appl. Phys. Lett. 2017, 111, 081101. [Google Scholar] [CrossRef]
- Kurita, T.; Mineyuki, N.; Shimotsuma, Y.; Fujiwara, M.; Mizuochi, N.; Shimizu, M.; Miura, K. Efficient generation of nitrogen-vacancy center inside diamond with shortening of laser pulse duration. Appl. Phys. Lett. 2018, 113, 211102. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic population analysis on LCAO–MO molecular wave functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef] [Green Version]
- Segall, M.D.; Shah, R.; Pickard, C.J.; Payne, M.C. Population analysis of plane-wave electronic structure calculations of bulk materials. Phys. Rev. B 1996, 54, 16317–16320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gali, A.; Maze, J.R. Ab initio study of the split silicon-vacancy defect in diamond: Electronic structure and related properties. Phys. Rev. B 2013, 88, 235205. [Google Scholar] [CrossRef] [Green Version]
- Petrone, A.; Beck, R.A.; Kasper, J.M.; Li, X.; Huang, Y.; Crane, M.; Pauzauskie, P. Electronic structures and spectroscopic signatures of silicon-vacancy containing nanodiamonds. Phys. Rev. B 2018, 98, 205405. [Google Scholar] [CrossRef] [Green Version]
- Kaviani, M.; Deák, P.; Aradi, B.; Frauenheim, T.; Chou, J.-P.; Gali, A. Proper surface termination for luminescent near-surface NV centers in diamond. Nano Lett. 2014, 14, 4772–4777. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H-Terminated | O-Terminated | N-Terminated | |||
|---|---|---|---|---|---|
| Site 1 | Site 2 | Site 1 | Site 2 | ||
| X–X | 1.62 | ||||
| X–C1 | 1.10 | 1.50 | 1.53 | ||
| C1–C1 | 1.62 | ||||
| C1–C2 | 1.54 | 1.54 | 1.52 | 1.59 | |
| C2–C3 | 1.53 | 1.57 | 1.54 | 1.52 | 1.57 |
| C3–C4 | 1.52 | 1.56 | 1.54 | 1.52 | 1.57 |
| C4–C5 | 1.52 | 1.56 | 1.54 | 1.54 | |
| C5–C6 | 1.54 | 1.54 | 1.55 | 1.53 | |
| C6–C7 | 1.55 | 1.54 | 1.54 | 1.54 | 1.54 |
| C7–C8 | 1.54 | 1.54 | 1.54 | 1.54 | 1.54 |
| H-terminated | O-terminated | N-terminated | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 1 | 2 | |||||||
| Si | V | Si | V | Si | V | Si | V | Si | V | |
| 2–3 | 1.95 | 4.30 | 3.79 | 2.68 | 6.55 | 4.83 | 7.80 | 3.81 | 3.63 | 5.26 |
| 3–4 | 4.92 | 4.52 | 3.32 | 5.31 | 5.13 | 5.47 | 6.27 | 5.84 | 3.75 | 4.58 |
| 4–5 | 5.61 | 5.03 | 3.74 | 4.22 | 5.59 | 5.27 | 5.29 | 5.01 | ||
| 5–6 | 4.88 | 4.99 | 5.08 | 5.20 | 5.06 | 4.79 | 5.42 | 5.62 | ||
| 6–7 | 4.70 | 4.53 | 5.13 | 5.12 | 5.27 | 5.22 | 4.94 | 5.15 | 5.31 | 5.24 |
| Bond | Bond Population | ||
|---|---|---|---|
| Bulk | Si–C | 1.98 | 0.54 |
| 0.55 | |||
| 0.57 | |||
| H-terminated | Si–C2 | 1.85 | 0.68 |
| Si–C3 | 2.02 | 0.65 | |
| Si–C4 | 1.94 | 0.70 | |
| Si–C5 | 2.37 | 0.22 | |
| O-terminated | Si–C1 | 2.08 | 0.45 |
| Si–C2 | 1.97 | 0.49 | |
| Si–C3 | 1.98 | 0.57 | |
| Si–C4 | 1.91 | 0.63 | |
| N-terminated | Si–C1 | 1.94 | 0.57 |
| Si–C2 | 2.01 | 0.54 | |
| Si–C3 | 1.96 | 0.64 | |
| Si–C4 | 2.19 | 0.40 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, Y.; Shen, W.; Shen, S.; Li, H. A Theoretical Study of the Energetic Stability and Geometry of Silicon-Vacancy Color Centers in Diamond (001) Surfaces. Appl. Sci. 2019, 9, 5471. https://doi.org/10.3390/app9245471
Pan Y, Shen W, Shen S, Li H. A Theoretical Study of the Energetic Stability and Geometry of Silicon-Vacancy Color Centers in Diamond (001) Surfaces. Applied Sciences. 2019; 9(24):5471. https://doi.org/10.3390/app9245471
Chicago/Turabian StylePan, Yuanhui, Wei Shen, Shengnan Shen, and Hui Li. 2019. "A Theoretical Study of the Energetic Stability and Geometry of Silicon-Vacancy Color Centers in Diamond (001) Surfaces" Applied Sciences 9, no. 24: 5471. https://doi.org/10.3390/app9245471